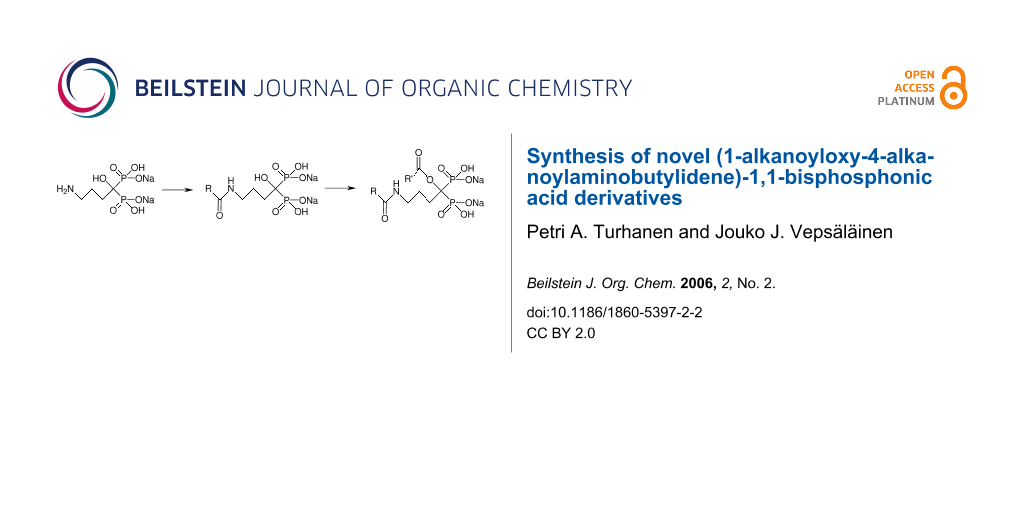
Abstract
A novel strategy for the synthesis of (1-alkanoyloxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives (1a-d) via (1-hydroxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives (2a-d), starting from alendronate has been developed with reasonable 51–77% overall yields. Intermediate products, (1-hydroxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives (2a-d), were prepared in water with reasonable to high yields (52–94%).

Graphical Abstract
Introduction
Bisphosphonates (BPs) are analogs of naturally occurring pyrophosphate and are used as drugs for the treatment of various bone diseases, like osteoporosis. [1-5] Alendronate, (1-hydroxy-4-aminobutylidene)-1,1-bisphosphonic acid (HABBPA) disodium salt is a typical example of an amino BP compound (Figure 1). Traditionally BPs have been used for decades as drugs for the treatment of various bone diseases but recently these compounds have been found to be active in many other fields, such as against parasitic diseases [6-10] and atherosclerosis. [11] Unfortunately BPs are very hydrophilic, highly anionic and their bioavailabilities are very poor. [12-14] It would be a clear advance to prepare more lipophilic and biodegradable derivatives of BPs. A rather straightforward method to improve the lipophilicity of amino BPs is to convert hydroxyl or/and amino group or the phosphonic acid groups with a biodegradable ester or amide functionality.
![[1860-5397-2-2-1]](/bjoc/content/figures/1860-5397-2-2-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our group has designed, synthesized and studied in vitro several different clodronate (R1 = R2 = Cl, Figure 1) and etidronate (R1 = CH3 R2 = OH, Figure 1) ester and amide derivatives to act as biodegradable prodrugs. [15-19] We have also recently described the method for the preparation of (1-alkoxycarbonyloxyethylidene)-1,1-bisphosphonic acid derivatives [20] and novel fatty acid derivatives of etidronate [21] which may represent prodrugs of etidronate. In the case of etidronic acid, we have shown that simple phosphonate esters like P(O)-O-Me as well as an acyl group at the tertiary alcohol group are stable against enzymatic hydrolysis. [18] However, the latter decomposed gradually due to chemical hydrolysis.
Because of the extreme hydrophilic character of HABBPA, its solubility and reactivity in organic solvents is very low. As far as we are aware there are only six known examples of amide (R-CO-HN-) derivatives of commercially used aminobisphosphonates (alendronate and pamidronate) in the literature, [22-24] but the hydroxyl derivatives are unknown. Probably one reason for this is the above mentioned solubility and reactivity problems.
Results and discussion
Here we report the first method to prepare (1-alkanoyloxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives 1a-d via (1-hydroxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid compounds 2a-d (see Scheme 1). The preparation of the intermediate compounds 2a-d was rather straightforward and was started from the mono sodium salt of HABBPA which was prepared and isolated as previously reported. [25] HABBPA mono sodium salt (200 mg) was stirred in water (2 ml) for a few minutes before a 40% NaOH solution (78 μl, 1.0 eq of NaOH) was added and the mixture was stirred until a clear solution was obtained. After addition of anhydride (1 ml), the mixture was stirred overnight (ca. 16 h for 2a-b and 24 h for 2c-d) and water was evaporated in vacuo, diethyl ether (10–15 ml) was added and the white precipitate was filtered and dried in vacuo. Since the products of 2a-d contained a few percent of the sodium salt of the corresponding carboxylic acid formed in the reaction and in the case of 2c, the reaction mixture contained 31% of starting material 3 (reaction did not proceed to completion in even 3 days) according to 1H and 31P NMR spectra, they required purification. Furthermore, in the case of 2a and 2b, 9% and 7% of 1a and 1b, respectively, were formed in the reaction. Compounds 2a-b were purified by dissolving in H2O (0.5 ml) and adding dry MeOH (3.5 ml) by stirring before the mixtures were placed in the freezer for 30 minutes. Solids were filtered and washed with H2O/MeOH (1:7) and dried in vacuo to produce 2a and 2b with 82% and 81% yields, respectively. Compound 2c was purified (if needed) by stirring in absolute EtOH for 30 minutes (this was repeated twice if necessary) to give 2c with 94% yield. Compound 2d was dissolved in water (3 ml) and dry MeOH (4 ml) was added by stirring before the mixture was placed in the freezer for weekend. The mixture was filtered and the filtrate was evaporated to dryness in vacuo to give 2d with a 52% yield. Alendronic acid (HABBPA), which was synthesized as reported earlier [25] (except for the isolation procedure, since in our hands, no precipitation occurred), was also tested as the starting material for the preparation of 2a (its acid form), but no reaction was observed under the test conditions.
![[1860-5397-2-2-i1]](/bjoc/content/inline/1860-5397-2-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of (1-alkanoyloxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives 1a (R = R' = Me), 1b (R = R' = Et), 1c (R = Pr; R' = Me) and 1d (R = But; R' = Me). Conditions: i) (RCO)2O in H2O; ii) (R'CO)2O then H2O (if necessary).
Scheme 1: Preparation of (1-alkanoyloxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives 1a (R =...
This is not an unexpected result since alendronic acid, which has pKa values of 0.8, 2.2, 6.3, 10.9 and 12.2 (Ezra et al. [26]), exists as a zwitterion (see Figure 1) and the ammonium group is protected against electrophilic attack of the acyl group.
Also the mono sodium salt of alendronic acid was a poor starting material due to the same reason but ca. 37% of 2a was obtained. As expected, the in situ prepared disodium salt of HABBPA was a good starting material because of its good solubility in water [solubility of HABBPA, its mono- and disodium salts in water (5 ml) were 41.4 mg, 161.7 mg and > 1000 mg, respectively, which were tested in room temperature] and reactivity of amino group when an excess of anhydride was used.
Since the amine group of alendronic acid is protected due to zwitterion, we attempted to acetylate the middle carbon OH-group directly by using alendronic acid and its monosodium salt under dry conditions, as we did for etidronic acid, [27] but unfortunately this did not prove feasible. However, acetylation was successful with this method when starting with the disodium salt of HABBPA. However, this led to a diacetylated product, with an acetyl moiety both, at the amino and the hydroxyl group.
Based on this finding the target compounds 1a-d were prepared from 2a-d using this method by stirring in an excess of acetic anhydride (1a,c,d) or propionic anhydride (1b). In the case of 1a and 1c, the reaction time was only 2 h and 8 h, respectively, whereas the reaction time for 1b and 1d was 48 h. We also tried to react compound 2c with butyric anhydride but the reaction did not occur under the test conditions. Acylation of OH-group with trimethylacetic anhydride was not successful due to steric hindrance of the trimethylacetic group. The target compounds 1a-d were isolated by filtration after addition of diethyl ether (10–15 ml) to the reaction mixture or by washing the residue with diethyl ether after the reaction mixture was evaporated to dryness. If needed, after drying the products were dissolved in a small volume of water and stirred for 40 minutes at room temperature for hydrolyzing any of formed bisphosphonate dimers which were obtained as determined by the 31P NMR spectra. Compounds 1a-c were purified like 2c by stirring in absolute EtOH, compound 1d needed no purification steps. All compounds (1a-d) still contained some sodium salt of the corresponding carboxylic acid (≤ 5%).
The formation of the intermediates 2a-d and the target compounds 1a-d during the reactions was readily confirmed from 1H and 31P NMR spectra. In 31P NMR spectra, the signals shift to the higher ppm value after the amino group had been acylated [e.g. for 3 from 18.59 ppm to 18.98 ppm (2a)]. However, after acylation of the OH-group, the 31P NMR signals shift to the lower ppm value [e.g. for 2a from 18.98 ppm to 14.65 ppm (1a)] as expected from the earlier results of the OH-group acylation from etidronic acid derivatives. [18,20,27]
Conclusion
In conclusion, the first synthesis of novel (1-alkanoyloxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives (1a-d) have been reported with good overall yields (51–77%) starting from alendronate. (1-Hydroxy-4-alkanoylaminobutylidene)-1,1-bisphosphonic acid derivatives (2a-d) were prepared and isolated with 52–94% yields as intermediate product for synthesis of 1a-d. All of the prepared compounds 1a-d and 2a-d are proposed to be more lipophilic and are thus more soluble in organic solvents than alendronate and can be used as starting materials for the synthesis of new phosphorus end modified derivatives of HABBPA and which may represent as possible prodrugs of alendronate. Novel biological evaluation for selected compounds of 1a-d and 2a-d will be carried out and published in future.
Supporting Information
| Supporting Information File 1: Experimental procedures and full spectroscopic data for all new compounds. | ||
| Format: DOC | Size: 31.5 KB | Download |
References
-
Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group, Inc.: New York, 1995.
Return to citation in text: [1] -
Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49.
Return to citation in text: [1] -
Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78.
Return to citation in text: [1] -
Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), 48S–52S. doi:10.1016/0002-9343(93)90383-Z
Return to citation in text: [1] -
Giannini, S.; D'Angelo, A.; L Sartori, L.; G Passeri, G.; Garbonare, L. D.; Crebaldi, C. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8
Return to citation in text: [1] -
Szajnman, S. H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3231–3235. doi:10.1016/S0960-894X(03)00663-2
Return to citation in text: [1] -
Szajnman, S. H.; Bailey, B. N.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2001, 11, 789–792. doi:10.1016/S0960-894X(01)00057-9
Return to citation in text: [1] -
Martin, M. B.; Sanders, J. M.; Kendrick, H.; Luca-Fradley, K.; Lewis, J. C.; Grimley, J. S.; Van Brussel, E. M.; Olsen, J. R.; Meints, G. A.; Burzynska, A.; Kafarski, P.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2002, 45, 2904–2914. doi:10.1021/jm0102809
Return to citation in text: [1] -
Garzoni, L. R.; Caldera, A.; Nazareth, L.; Meirelles, M.; Castro, S. L.; Docampo, R.; Meints, G. A.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 273–285. doi:10.1016/j.ijantimicag.2003.07.020
Return to citation in text: [1] -
Garzoni, L. R.; Waghabi, M. C.; Baptista, M. M.; Castro, S. L.; Nazareth, L.; Meirelles, M.; Britto, C. C.; Docampo, R.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 286–290. doi:10.1016/j.ijantimicag.2003.07.019
Return to citation in text: [1] -
Ylitalo, R. Gen. Pharmacol. 2002, 35, 287–296.
Return to citation in text: [1] -
Vepsäläinen, J. J. Curr. Med. Chem. 2002, 9, 1201–1208.
Return to citation in text: [1] -
Lin, J. H. Bone 1996, 18, 75–85. doi:10.1016/8756-3282(95)00445-9
Return to citation in text: [1] -
Recker, R. R.; Saville, P. D. Toxicol. Appl. Pharmacol. 1973, 24, 580–589. doi:10.1016/0041-008X(73)90219-6
Return to citation in text: [1] -
Turhanen, P. A.; Niemi, R.; Peräkylä, M.; Järvinen, T.; Vepsäläinen, J. J. Org. Biomol. Chem. 2003, 1, 3223–3226. doi:10.1039/b305979k
Return to citation in text: [1] -
Ahlmark, M.; Vepsäläinen, J.; Taipale, H.; Niemi, R.; Järvinen, T. J. Med. Chem. 1999, 42, 1473–1476. doi:10.1021/jm9810809
Return to citation in text: [1] -
Niemi, R.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. J. Med. Chem. 1999, 42, 5053–5058. doi:10.1021/jm991109o
Return to citation in text: [1] -
Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3
Return to citation in text: [1] [2] [3] -
Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 4, 633–637. doi:10.1055/s-2001-12353
Return to citation in text: [1] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 13, 2119–2121. doi:10.1055/s-2005-869984
Return to citation in text: [1] [2] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 18, 3063–3066. doi:10.1055/s-2005-916032
Return to citation in text: [1] -
Kofi Adzamli, I.; Gries, H.; Johnson, D.; Blau, M. J. Med. Chem. 1989, 32, 139–144. doi:10.1021/jm00121a026
Return to citation in text: [1] -
Ezra, A.; Hoffman, A.; Breuer, E.; Alferiev, I. S.; Mönkkönen, J.; Hanany-Rozen, N. E.; Weiss, G.; Stepensky, D.; Gati, I.; Cohen, H.; Törmälehto, S.; Amidon, G. L.; Golomb, G. J. Med. Chem. 2000, 43, 3641–3652. doi:10.1021/jm980645y
Return to citation in text: [1] -
Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmid, J.; Scheiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i
Return to citation in text: [1] -
Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.; Grenda, V. J.; Shinkai, I. J. Org. Chem. 1995, 60, 8310–8312. doi:10.1021/jo00130a036
Return to citation in text: [1] [2] -
Ezra, A.; Golomb, G. Adv. Drug Delivery Rev. 2000, 42, 175. doi:10.1016/S0169-409X(00)00061-2
Return to citation in text: [1] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 7, 992–994. doi:10.1055/s-2004-822345
Return to citation in text: [1] [2]
| 1. | Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group, Inc.: New York, 1995. |
| 2. | Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49. |
| 3. | Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78. |
| 4. | Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), 48S–52S. doi:10.1016/0002-9343(93)90383-Z |
| 5. | Giannini, S.; D'Angelo, A.; L Sartori, L.; G Passeri, G.; Garbonare, L. D.; Crebaldi, C. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8 |
| 15. | Turhanen, P. A.; Niemi, R.; Peräkylä, M.; Järvinen, T.; Vepsäläinen, J. J. Org. Biomol. Chem. 2003, 1, 3223–3226. doi:10.1039/b305979k |
| 16. | Ahlmark, M.; Vepsäläinen, J.; Taipale, H.; Niemi, R.; Järvinen, T. J. Med. Chem. 1999, 42, 1473–1476. doi:10.1021/jm9810809 |
| 17. | Niemi, R.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. J. Med. Chem. 1999, 42, 5053–5058. doi:10.1021/jm991109o |
| 18. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 19. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 4, 633–637. doi:10.1055/s-2001-12353 |
| 12. | Vepsäläinen, J. J. Curr. Med. Chem. 2002, 9, 1201–1208. |
| 13. | Lin, J. H. Bone 1996, 18, 75–85. doi:10.1016/8756-3282(95)00445-9 |
| 14. | Recker, R. R.; Saville, P. D. Toxicol. Appl. Pharmacol. 1973, 24, 580–589. doi:10.1016/0041-008X(73)90219-6 |
| 27. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 7, 992–994. doi:10.1055/s-2004-822345 |
| 6. | Szajnman, S. H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3231–3235. doi:10.1016/S0960-894X(03)00663-2 |
| 7. | Szajnman, S. H.; Bailey, B. N.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2001, 11, 789–792. doi:10.1016/S0960-894X(01)00057-9 |
| 8. | Martin, M. B.; Sanders, J. M.; Kendrick, H.; Luca-Fradley, K.; Lewis, J. C.; Grimley, J. S.; Van Brussel, E. M.; Olsen, J. R.; Meints, G. A.; Burzynska, A.; Kafarski, P.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2002, 45, 2904–2914. doi:10.1021/jm0102809 |
| 9. | Garzoni, L. R.; Caldera, A.; Nazareth, L.; Meirelles, M.; Castro, S. L.; Docampo, R.; Meints, G. A.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 273–285. doi:10.1016/j.ijantimicag.2003.07.020 |
| 10. | Garzoni, L. R.; Waghabi, M. C.; Baptista, M. M.; Castro, S. L.; Nazareth, L.; Meirelles, M.; Britto, C. C.; Docampo, R.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 286–290. doi:10.1016/j.ijantimicag.2003.07.019 |
| 18. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 20. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 13, 2119–2121. doi:10.1055/s-2005-869984 |
| 27. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 7, 992–994. doi:10.1055/s-2004-822345 |
| 22. | Kofi Adzamli, I.; Gries, H.; Johnson, D.; Blau, M. J. Med. Chem. 1989, 32, 139–144. doi:10.1021/jm00121a026 |
| 23. | Ezra, A.; Hoffman, A.; Breuer, E.; Alferiev, I. S.; Mönkkönen, J.; Hanany-Rozen, N. E.; Weiss, G.; Stepensky, D.; Gati, I.; Cohen, H.; Törmälehto, S.; Amidon, G. L.; Golomb, G. J. Med. Chem. 2000, 43, 3641–3652. doi:10.1021/jm980645y |
| 24. | Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmid, J.; Scheiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i |
| 25. | Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.; Grenda, V. J.; Shinkai, I. J. Org. Chem. 1995, 60, 8310–8312. doi:10.1021/jo00130a036 |
| 18. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 26. | Ezra, A.; Golomb, G. Adv. Drug Delivery Rev. 2000, 42, 175. doi:10.1016/S0169-409X(00)00061-2 |
| 21. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 18, 3063–3066. doi:10.1055/s-2005-916032 |
| 20. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 13, 2119–2121. doi:10.1055/s-2005-869984 |
| 25. | Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.; Grenda, V. J.; Shinkai, I. J. Org. Chem. 1995, 60, 8310–8312. doi:10.1021/jo00130a036 |
© 2006 Turhanen and Vepsäläinen; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)