Abstract
The use of organophosphorus derivatising agents, prepared from C2 symmetric trimethylsilylated diamines, for the 1H NMR and 31P NMR determination of the enantiomeric composition of chiral alcohols is described.

Graphical Abstract
Introduction
NMR spectroscopy is one of the most frequently employed methods used for determining the enantiomeric purity of chiral compounds, based on the formation of diastereomeric complexes or derivatives [1]. Among these methods, 31P is a very attractive nucleus to be used for NMR analysis because of the large chemical dispersion and the simplicity of the spectra [2]. Some of the chiral phosphorous chemical derivatisation agents (CDAs) developed contain an amine or a C2 symmetric diamine moiety, and have conveniently been applied to the determination of the enantiomeric excess (ee) of various chiral alcohols, [3-20], amines,[6,13,14,21,22] thiols[3,10,12,13,22,23] and amino acids[21,24,25]. Analyses are also sometimes made by 1H or 19F NMR [26]. This is the case with α-methoxy-α-(trifluoromethyl)phenylacetic acid (Mosher's, MTPA) esters[27] and other arylmethoxyacetic acid derivatives for the determination of enantiomeric excesses and attribution of configuration of certain alcohols, amines and carboxylic acids. The scope and limitations of these methods have been discussed by Seco et al [28]. However, the analysis of the 1H NMR spectra is often more complicated than that of 31P NMR spectra because of the H-H coupling, the lower resolution and the superposition of the desired signals with other proton signals. We therefore decided to develop new CDAs to determine the enantiomeric composition by a simple 1H NMR method.
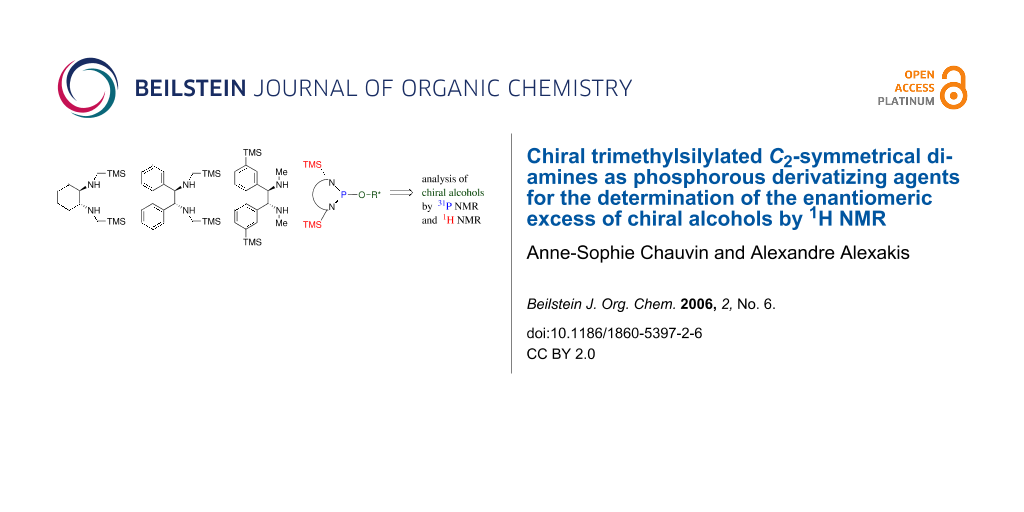
To be a convenient method, the signal to be observed should belong to an area usually devoid of any other signals, which can allow the determination of ee by integration. The area around 0 ppm usually fulfils these criteria. We already reported the use of TADDOL phosphorus derivatives, where the determination of ee can be performed by integration of the TADDOL isopropylidene-methyl signals [29]. Herein we report the use of CDAs 1, 2 and 3, obtained from C2 symmetric trimethylsilylated diamines (Scheme 1), for the determination of the enantiomeric composition of chiral alcohols, including phenylcarbinols, not only by 31P NMR but also by 1H NMR. The choice of the position of the TMS group was essential, to see if it is more discriminating on the nitrogen atom or on the backbone.
![[1860-5397-2-6-i23]](/bjoc/content/inline/1860-5397-2-6-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C2-diamines used in this study.
Scheme 1: C2-diamines used in this study.
Results and discussion
(R,R)-N,N'-Bis-trimethylsilanylmethylcyclohexane-1,2-diamine 1 was synthesised starting from the stable (R,R)-1,2-diammoniumcyclohexane mono-(+)-tartrate salt 4 and chloromethyltrimethylsilane (Scheme 2), according to a described procedure [30]. (R,R)-1,2-diphenyl-N,N'-bis-trimethylsilanylmethyl-ethane-1,2-diamine 2 was prepared by the same method, starting from (R,R)-diphenyl-ethane-1,2-diamine 5 (Scheme 2) [31]. N,N'-Dimethyl-1,2-bis-(3-trimethylsilanyl-phenyl)-ethane-1,2-diamine 3 was synthesised by reductive coupling of methyl-(3-trimethylsilanylbenzylidene)imine 8, which was synthesised in a three step procedure according to Scheme 3. Only the racemic diamine was evaluated for the chemical shift difference Δδ by 31P and 1H NMR. All experimental details are given in Supporting Information File 1.
![[1860-5397-2-6-i24]](/bjoc/content/inline/1860-5397-2-6-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-2-6-i25]](/bjoc/content/inline/1860-5397-2-6-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to increase the 31P chemical shift differences of P(III) derivatives in the presence of chiral alcohols, it was found that the best position for substituting the phenyl ring of the C2 symmetrical diamines is meta compared to ortho or para position [32]. It is for this reason that we introduced the methyltrimethylsilyl substituent in the meta position of diamine 3 (Scheme 3).
With butan-2-ol taken as representative example, a comparison of the 31P chemical shift differences Δδ (ppm) of some C2 symmetrical diamine-P(III) derivatives is presented in Table 1. The Δδ between the two diastereoisomers is of the same magnitude when the nitrogen atom is substituted by a methyl or a methyltrimethylsilyl moiety, despite the latter being a more bulky substituent (9 and 10, or 11 and 12). However, the Δδ value increases by a factor of two in the case of cyclohexane-1,2-diamine derivatives (9 and 10) compared to 1,2-diphenyl-ethane-1,2-diamine derivatives (11 to 13) and can be explained by the rigidity of the cyclohexane ring. In the case of 11 and 13, the chemical shift difference is roughly the same, indicating that the introduction of the trimethylsilyl moiety in the aromatic ring has more or less no influence. In all cases, the resolution of both diastereoisomeric peaks is large enough to allow the determination of ee by integration of their corresponding areas.
![[Graphic 1]](/bjoc/content/inline/1860-5397-2-6-i1.svg?max-width=637&scale=0.9690924)
![[Graphic 2]](/bjoc/content/inline/1860-5397-2-6-i2.svg?max-width=637&scale=0.9690924)
![[Graphic 3]](/bjoc/content/inline/1860-5397-2-6-i3.svg?max-width=637&scale=0.9690924)
![[Graphic 4]](/bjoc/content/inline/1860-5397-2-6-i4.svg?max-width=637&scale=0.9690924)
![[Graphic 5]](/bjoc/content/inline/1860-5397-2-6-i5.svg?max-width=637&scale=0.9690924)
More interesting is the application of these silylated compounds for the direct determination of the ee by 1H NMR. The chemical shift δ (in ppm) of two diastereoisomers obtained from the same C2 symmetrical diamine-P(III) derivatives and a chiral alcohol are presented in Table 2. For each diastereoisomer, two singlets are observed in the region around 0 ppm, due to the two non-equivalent trimethysilyl substituents (see Figure 1). No other signal can interfere in this region. Futhermore, these signals are very intense, due to the presence of nine equivalent protons for the three methyls of each trimethysilyl substituent. As observed in Table 2, the resolution of the signals is good enough, so that the determination of the enantiomeric excess by integration of each diastereoisomer peak can be easily performed. This ee determination can be made either with the P(III) or with the P(V) derivatives, after reaction with sulphur, although superposition of peaks occurs more often in the latter case. With some N,N'-dimethyl-1,2-bis-(3-trimethylsilanyl-phenyl)-ethane-1,2-diamine 3 derivatives, especially the P(V) ones, the resolution of the two trimethylsilyl groups of one diastereoisomer decreases or does not exist, probably due to the compound's greater symmetry, but the separation between each diastereoisomer remains very good. In such a case, it is better to integrate both areas of peaks belonging to the same diastereoisomers. The ee values have been found to be the same (within experimental errors) when measured by integration of 31P or of 1H peaks (for both P(III) or P(V) derivatives). Interestingly, in the examples studied, the chemical shift difference Δδ between the two peaks of the diastereoisomer arising from the (R,R)-C2-diamine and the (R)-alcohol is often higher than that arising from the (S,S)-C2-diamine. This observation could on occasion be considered for the assignment of the absolute configuration of these chiral alcohols.
Table 2: Chemical shifts of the methyls of trimethylsilyls substituents.
![[Graphic 6]](/bjoc/content/inline/1860-5397-2-6-i6.svg?max-width=637&scale=0.9690924)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-2-6-i7.svg?max-width=637&scale=0.9690924)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-2-6-i8.svg?max-width=637&scale=0.9690924)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-2-6-i9.svg?max-width=637&scale=0.9690924)
|
|||||
| (R,R) | (S,S) | (R,R) | (S,S) | |||||
|---|---|---|---|---|---|---|---|---|
| δ (ppm) | δ (ppm) | δ (ppm) | δ (ppm) | |||||
![[Graphic 10]](/bjoc/content/inline/1860-5397-2-6-i10.svg?max-width=637&scale=1.0)
|
+0.12 | -0.06 | +0.06 | -0.02 | 0.08 | -0.10 | 0.11 | -0.05 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-2-6-i11.svg?max-width=637&scale=1.0)
|
0.09 | -0.03 | 0.07 | -0.00 | 0.09 | -0.08 | 0.12 | -0.11 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-2-6-i12.svg?max-width=637&scale=1.0)
|
0.14 | -0.05 | -0.03 | -0.12 | 0.09 | -0.06 | 0.10 | -0.09 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-2-6-i13.svg?max-width=637&scale=1.0)
|
0.26 | -0.06 | 0.12 | 0.10 | 0.10 | 0.06 | 0.08 | 0.11 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-2-6-i14.svg?max-width=637&scale=0.9690924)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-2-6-i15.svg?max-width=637&scale=0.9690924)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-2-6-i16.svg?max-width=637&scale=0.9690924)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-2-6-i17.svg?max-width=637&scale=0.9690924)
|
|||||
| (R,R) | (S,S) | (R,R) | (S,S) | |||||
|
|
+0.11 | -0.04 | +0.09 | -0.10 | -0.04 | -0.10 | -0.11 | -0.39 |
|
|
0.01 | -0.06 | 0.08 | -0.17 | -0.16 | -0.28 | -0.11 | -0.41 |
|
|
0.09 | -0.07 | 0.10 | -0.14 | 0.06 | -0.29 | -0.11 | -0.39 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-2-6-i18.svg?max-width=637&scale=1.0)
|
0.05 | -0.03 | 0.06 | -0.03 | +0.11 | -0.12 | 0.02 | -0.03 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-2-6-i19.svg?max-width=637&scale=0.9690924)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-2-6-i20.svg?max-width=637&scale=0.9690924)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-2-6-i21.svg?max-width=637&scale=0.9690924)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-2-6-i22.svg?max-width=637&scale=0.9690924)
|
|||||
| (R,R) | (S,S) | (R,R) | (S,S) | |||||
|
|
0.15 | 0.14 | 0.11 | 0.10 | 0.15 | 0.11 | 0.10 | 0.09 |
|
|
0.13 | 0.11 | 0.09 | 0.08 | 0.13 | 0.12 | 0.10 | 0.08 |
|
|
0.12 | 0.09 | 0.08 | 0.06 | 0.15 | 0.15 | 0.03 | 0.00 |
|
|
0.12 | 0.10 | 0.08 | 0.05 | 0.14 | 0.14 | 0.02 | 0.01 |
![[1860-5397-2-6-1]](/bjoc/content/figures/1860-5397-2-6-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectrum in the -0.4↔0.6 ppm region of 2-(2-isopropyl-5-methyl-cyclohexyloxy)-1,3-bis-trimethylsilanylmethyl-octahydro-benzo [1,3,2] diazaphosphole 2-sulfide.
Figure 1: 1H NMR spectrum in the -0.4↔0.6 ppm region of 2-(2-isopropyl-5-methyl-cyclohexyloxy)-1,3-bis-trimet...
Conclusion
In conclusion, we have synthesized three new C2 symmetric silylated diamines 1, 2 and 3, which can be easily used as CDAs for the 1H NMR and 31P NMR determination of the enantiomeric composition of secondary chiral alcohols, including phenylcarbinols. Diamines 1 and 2 are the most suitable for such a determination, whereas diamine 3 affords a lower Δδ. It seems that the position of the silyl derivative is very important: it is clear that the TMS group has to be closer to the stereocenter of the alcohol, although there is no effect due to the presence of a cyclohexyl ring instead of two aromatic substituents. If one takes into account the ease of preparation of these diamines, then, (R,R)-N,N'-bis-trimethylsilanylmethyl-cyclohexane-1,2-diamine 1 would be preferred. Work is in progress to extend this methodology to the determination of the absolute configuration of chiral alcohols, as has been done with 31P NMR [33].
Supporting Information
| Supporting Information File 1: Contains all experimental data | ||
| Format: DOC | Size: 37.5 KB | Download |
References
-
Wenzel, T.; Wilcox, J. D. Chirality 2003, 15, 256. doi:10.1002/chir.10190
and references therein.
Return to citation in text: [1] -
Verkade, J. G.; Quin, L. D. Phosphorus 31 NMR spectroscopy in stereochemical analysis: organic compounds and metal complexes; Methods in stereochemical Analysis, Vol. 8; VCH publishers, Inc.: Deerfield Beach, Florida, 1987.
Return to citation in text: [1] -
Feringa, B. L.; Smaardijk, A. A.; Wynberg, H.; Strijtveen, B.; Kellogg, R. M. Tetrahedron Lett. 1986, 27, 997–1000. doi:10.1016/S0040-4039(00)84160-X
Return to citation in text: [1] [2] -
Brunel, J.-M.; Pardigon, O.; Maffrei, M.; Buono, G. Tetrahedron: Asymmetry 1992, 3, 1243. doi:10.1016/S0957-4166(00)82082-0
Return to citation in text: [1] -
Anderson, R. C.; Shapiro, M. J. J. Org. Chem. 1984, 49, 1304. doi:10.1021/jo00181a042
Return to citation in text: [1] -
Jonhson, C. R.; Elliott, R. C.; Penning, T. D. J. Am. Chem. Soc. 1984, 106, 5019. doi:10.1021/ja00329a072
Return to citation in text: [1] [2] -
Feringa, B. L.; Smaardijk, A.; Wynberg, H. J. Am. Chem. Soc. 1985, 107, 4798–4799. doi:10.1021/ja00302a043
Return to citation in text: [1] -
Feringa, B. L. J. Chem. Soc., Chem. Commun. 1987, 695. doi:10.1039/c39870000695
Return to citation in text: [1] -
Kato, N. J. Am. Chem. Soc. 1990, 112, 254. doi:10.1021/ja00157a039
Return to citation in text: [1] -
Alexakis, A.; Mutti, S.; Normant, J. F.; Mangeney, P. Tetrahedron: Asymmetry 1990, 1, 437. doi:10.1016/S0957-4166(00)86347-8
Return to citation in text: [1] [2] -
Welch, J. C. Tetrahedron: Asymmetry 1991, 2, 1127. doi:10.1016/S0957-4166(00)82011-X
Return to citation in text: [1] -
Alexakis, A.; Mutti, S.; Mangeney, P. J. Org. Chem. 1992, 57, 1224. doi:10.1021/jo00030a034
Return to citation in text: [1] [2] -
Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. Tetrahedron Lett. 1994, 35, 5125. doi:10.1016/S0040-4039(00)77044-4
Return to citation in text: [1] [2] [3] -
Oshikawa, T.; Yamashita, M.; Kumugai, S.; Seo, K.; Kobayashi, J. J. Chem. Soc., Chem. Commun. 1995, 435. doi:10.1039/c39950000435
Return to citation in text: [1] [2] -
Brunel, J. M.; Faure, B. Tetrahedron: Asymmetry 1995, 6, 2353–2356. doi:10.1016/0957-4166(95)00311-C
Return to citation in text: [1] -
Garner, C. M.; McWhorter, C.; Goerke, A. R. Tetrahedron Lett. 1997, 38, 7717. doi:10.1016/S0040-4039(97)10064-8
Return to citation in text: [1] -
de Parrodi, C. A.; Moreno, G. E.; Quintero, L.; Juaristi, E. Tetrahedron: Asymmetry 1998, 9, 2093. doi:10.1016/S0957-4166(98)00219-5
Return to citation in text: [1] -
Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. J. Org. Chem. 1994, 59, 3326. doi:10.1021/jo00091a019
Return to citation in text: [1] -
Devitt, P. G.; Mitchell, M. C.; Weetman, J. M.; Taylor, R. J.; Kee, T. P. Tetrahedron: Asymmetry 1995, 6, 2039. doi:10.1016/0957-4166(95)00265-Q
Return to citation in text: [1] -
Reymond, S.; Brunel, J. M.; Buono, G. Tetrahedron: Asymmetry 2000, 11, 1273. doi:10.1016/S0957-4166(00)00062-8
Return to citation in text: [1] -
Feringa, B. L.; Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 5484–5486. doi:10.1021/jo00376a100
Return to citation in text: [1] [2] -
Kolodiazhnyi, O. I.; Demchuk, O. M.; Gerschkovich, A. A. Tetrahedron: Asymmetry 1999, 10, 1729–1732. doi:10.1016/S0957-4166(99)00139-1
Return to citation in text: [1] [2] -
Strijtveen, B.; Feringa, B. L.; Kellogg, R. M. Tetrahedron 1987, 43, 123–130. doi:10.1016/S0040-4020(01)89938-5
Return to citation in text: [1] -
Hulst, R.; de Vries, N. K.; Feringa, B. L. Angew. Chem., Int. Ed. Engl. 1992, 31, 1093. doi:10.1002/anie.199210921
Return to citation in text: [1] -
Hulst, R.; Zijlstra, R. W. J.; de Vries, N. K.; Feringa, B. L. Tetrahedron: Asymmetry 1994, 5, 1701–1710. doi:10.1016/0957-4166(94)80081-2
Return to citation in text: [1] -
Parker, D. Chem. Rev. 1991, 91, 1441. doi:10.1021/cr00007a009
Return to citation in text: [1] -
Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512. doi:10.1021/ja00783a034
Return to citation in text: [1] -
Seco, J. M.; Quinoa, E.; Riguera, R. Tetrahedron: Asymmetry 2001, 12, 2915. doi:10.1016/S0957-4166(01)00508-0
Return to citation in text: [1] -
Alexakis, A.; Chauvin, A.-S. Tetrahedron: Asymmetry 2001, 11, 4245. doi:10.1016/S0957-4166(00)00400-6
Return to citation in text: [1] -
Alexakis, A.; Chauvin, A.-S.; Stouvenel, R.; Vrancken, E.; Mutti, S.; Mangeney, P. Tetrahedron: Asymmetry 2001, 12, 1171. doi:10.1016/S0957-4166(01)00198-7
Return to citation in text: [1] -
Pikul, S.; Corey, E. J. Org. Synth. 1993, 71, 22.
Return to citation in text: [1] -
Cuvinot, D.; Mangeney, P.; Alexakis, A.; Normant, J. F.; Lellouche, J. P. J. Org. Chem. 1989, 54, 2420. doi:10.1021/jo00271a034
Return to citation in text: [1] -
Chauvin, A.-S.; Bernardinelli, G.; Alexakis, A. Tetrahedron: Asymmetry 2004, 15, 1857–1879. doi:10.1016/j.tetasy.2004.04.031
Return to citation in text: [1]
| 1. |
Wenzel, T.; Wilcox, J. D. Chirality 2003, 15, 256. doi:10.1002/chir.10190
and references therein. |
| 3. | Feringa, B. L.; Smaardijk, A. A.; Wynberg, H.; Strijtveen, B.; Kellogg, R. M. Tetrahedron Lett. 1986, 27, 997–1000. doi:10.1016/S0040-4039(00)84160-X |
| 10. | Alexakis, A.; Mutti, S.; Normant, J. F.; Mangeney, P. Tetrahedron: Asymmetry 1990, 1, 437. doi:10.1016/S0957-4166(00)86347-8 |
| 12. | Alexakis, A.; Mutti, S.; Mangeney, P. J. Org. Chem. 1992, 57, 1224. doi:10.1021/jo00030a034 |
| 13. | Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. Tetrahedron Lett. 1994, 35, 5125. doi:10.1016/S0040-4039(00)77044-4 |
| 22. | Kolodiazhnyi, O. I.; Demchuk, O. M.; Gerschkovich, A. A. Tetrahedron: Asymmetry 1999, 10, 1729–1732. doi:10.1016/S0957-4166(99)00139-1 |
| 23. | Strijtveen, B.; Feringa, B. L.; Kellogg, R. M. Tetrahedron 1987, 43, 123–130. doi:10.1016/S0040-4020(01)89938-5 |
| 6. | Jonhson, C. R.; Elliott, R. C.; Penning, T. D. J. Am. Chem. Soc. 1984, 106, 5019. doi:10.1021/ja00329a072 |
| 13. | Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. Tetrahedron Lett. 1994, 35, 5125. doi:10.1016/S0040-4039(00)77044-4 |
| 14. | Oshikawa, T.; Yamashita, M.; Kumugai, S.; Seo, K.; Kobayashi, J. J. Chem. Soc., Chem. Commun. 1995, 435. doi:10.1039/c39950000435 |
| 21. | Feringa, B. L.; Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 5484–5486. doi:10.1021/jo00376a100 |
| 22. | Kolodiazhnyi, O. I.; Demchuk, O. M.; Gerschkovich, A. A. Tetrahedron: Asymmetry 1999, 10, 1729–1732. doi:10.1016/S0957-4166(99)00139-1 |
| 3. | Feringa, B. L.; Smaardijk, A. A.; Wynberg, H.; Strijtveen, B.; Kellogg, R. M. Tetrahedron Lett. 1986, 27, 997–1000. doi:10.1016/S0040-4039(00)84160-X |
| 4. | Brunel, J.-M.; Pardigon, O.; Maffrei, M.; Buono, G. Tetrahedron: Asymmetry 1992, 3, 1243. doi:10.1016/S0957-4166(00)82082-0 |
| 5. | Anderson, R. C.; Shapiro, M. J. J. Org. Chem. 1984, 49, 1304. doi:10.1021/jo00181a042 |
| 6. | Jonhson, C. R.; Elliott, R. C.; Penning, T. D. J. Am. Chem. Soc. 1984, 106, 5019. doi:10.1021/ja00329a072 |
| 7. | Feringa, B. L.; Smaardijk, A.; Wynberg, H. J. Am. Chem. Soc. 1985, 107, 4798–4799. doi:10.1021/ja00302a043 |
| 8. | Feringa, B. L. J. Chem. Soc., Chem. Commun. 1987, 695. doi:10.1039/c39870000695 |
| 9. | Kato, N. J. Am. Chem. Soc. 1990, 112, 254. doi:10.1021/ja00157a039 |
| 10. | Alexakis, A.; Mutti, S.; Normant, J. F.; Mangeney, P. Tetrahedron: Asymmetry 1990, 1, 437. doi:10.1016/S0957-4166(00)86347-8 |
| 11. | Welch, J. C. Tetrahedron: Asymmetry 1991, 2, 1127. doi:10.1016/S0957-4166(00)82011-X |
| 12. | Alexakis, A.; Mutti, S.; Mangeney, P. J. Org. Chem. 1992, 57, 1224. doi:10.1021/jo00030a034 |
| 13. | Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. Tetrahedron Lett. 1994, 35, 5125. doi:10.1016/S0040-4039(00)77044-4 |
| 14. | Oshikawa, T.; Yamashita, M.; Kumugai, S.; Seo, K.; Kobayashi, J. J. Chem. Soc., Chem. Commun. 1995, 435. doi:10.1039/c39950000435 |
| 15. | Brunel, J. M.; Faure, B. Tetrahedron: Asymmetry 1995, 6, 2353–2356. doi:10.1016/0957-4166(95)00311-C |
| 16. | Garner, C. M.; McWhorter, C.; Goerke, A. R. Tetrahedron Lett. 1997, 38, 7717. doi:10.1016/S0040-4039(97)10064-8 |
| 17. | de Parrodi, C. A.; Moreno, G. E.; Quintero, L.; Juaristi, E. Tetrahedron: Asymmetry 1998, 9, 2093. doi:10.1016/S0957-4166(98)00219-5 |
| 18. | Alexakis, A.; Frutos, J. C.; Mutti, S.; Mangeney, P. J. Org. Chem. 1994, 59, 3326. doi:10.1021/jo00091a019 |
| 19. | Devitt, P. G.; Mitchell, M. C.; Weetman, J. M.; Taylor, R. J.; Kee, T. P. Tetrahedron: Asymmetry 1995, 6, 2039. doi:10.1016/0957-4166(95)00265-Q |
| 20. | Reymond, S.; Brunel, J. M.; Buono, G. Tetrahedron: Asymmetry 2000, 11, 1273. doi:10.1016/S0957-4166(00)00062-8 |
| 32. | Cuvinot, D.; Mangeney, P.; Alexakis, A.; Normant, J. F.; Lellouche, J. P. J. Org. Chem. 1989, 54, 2420. doi:10.1021/jo00271a034 |
| 2. | Verkade, J. G.; Quin, L. D. Phosphorus 31 NMR spectroscopy in stereochemical analysis: organic compounds and metal complexes; Methods in stereochemical Analysis, Vol. 8; VCH publishers, Inc.: Deerfield Beach, Florida, 1987. |
| 33. | Chauvin, A.-S.; Bernardinelli, G.; Alexakis, A. Tetrahedron: Asymmetry 2004, 15, 1857–1879. doi:10.1016/j.tetasy.2004.04.031 |
| 28. | Seco, J. M.; Quinoa, E.; Riguera, R. Tetrahedron: Asymmetry 2001, 12, 2915. doi:10.1016/S0957-4166(01)00508-0 |
| 30. | Alexakis, A.; Chauvin, A.-S.; Stouvenel, R.; Vrancken, E.; Mutti, S.; Mangeney, P. Tetrahedron: Asymmetry 2001, 12, 1171. doi:10.1016/S0957-4166(01)00198-7 |
| 27. | Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512. doi:10.1021/ja00783a034 |
| 21. | Feringa, B. L.; Strijtveen, B.; Kellogg, R. M. J. Org. Chem. 1986, 51, 5484–5486. doi:10.1021/jo00376a100 |
| 24. | Hulst, R.; de Vries, N. K.; Feringa, B. L. Angew. Chem., Int. Ed. Engl. 1992, 31, 1093. doi:10.1002/anie.199210921 |
| 25. | Hulst, R.; Zijlstra, R. W. J.; de Vries, N. K.; Feringa, B. L. Tetrahedron: Asymmetry 1994, 5, 1701–1710. doi:10.1016/0957-4166(94)80081-2 |
| 29. | Alexakis, A.; Chauvin, A.-S. Tetrahedron: Asymmetry 2001, 11, 4245. doi:10.1016/S0957-4166(00)00400-6 |
© 2006 Chauvin and Alexakis; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)