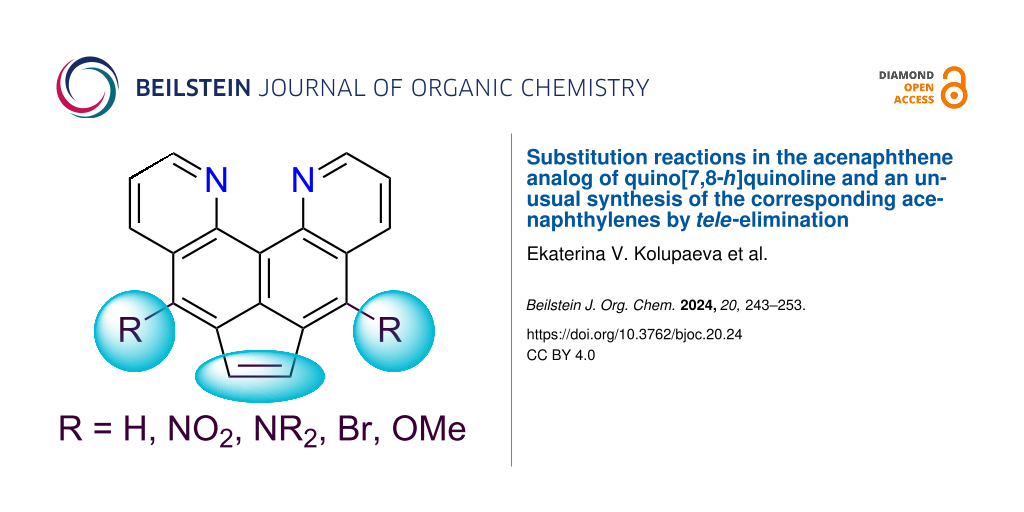
Abstract
The possibility of functionalization of dipyrido[3,2-e:2′,3′-h]acenaphthene containing a quino[7,8-h]quinoline fragment and being a highly basic diazine analog of 1,8-bis(dimethylamino)naphthalene (“proton sponge”) has been studied for the first time. In addition to the pronounced tendency of the title compound to form associates with an intramolecular hydrogen bond of the NHN type (new examples with the participation of pyridine rings, including self-associates are shown) and its inertness to amination reactions of the pyridine rings, the naphthalene core at positions 5(8) and the CH2CH2 bridge (dehydrogenation) undergo chemical modifications under mild conditions, giving the corresponding acenaphthylenes. The latter can also be obtained in an unusual way by tele-elimination from 5,8-dibromodipyridoacenaphthene by reaction with neutral or anionic bases.

Graphical Abstract
Introduction
Quinoline derivatives, classical nitrogen-containing heterocycles, are widely distributed in nature in various forms and used in medicine, food industry, catalysts, dyes, functional materials, oil refining, and electronics [1,2]. Quinoline and its derivatives have antibiotic, antimalarial, antitumor, anti-inflammatory, antihypertensive, and antiretroviral properties [3,4]. Therefore, at present, there is a need for compounds containing a quinoline fragment in various fields of research.
At the same time, quinoline bases are a popular platform for the molecular design of polycyclic systems with receptor properties; they easily form proton complexes with high stability and selectivity [5,6]. This, in turn, attracts the attention of researchers involved in the study of various types of hydrogen bonds and the problem of superbasicity [7,8]. Indeed, the basicity of quinoline and simple azaarenes is rather low. At the same time, the correct structural organization of azaarenes, where unshared electron pairs are forced to strongly repel each other in space, can lead to a sharp increase in basicity [8]. Thus, quino[7,8-h]quinoline (3), first obtained in the Staab group [9], already exceeds in basicity not only quinoline itself, but also 1,8-bis(dimethylamino)naphthalene (1, “proton sponge”) (Scheme 1; pKa values of the corresponding monoprotonated forms are given). Meanwhile, the very synthesis of polycondensed quinoline bases with a certain arrangement of nitrogen atoms is often a challenge for a synthetic scientist [9,10]. This greatly limits the possible use of such polynuclear azaarenes in organic synthesis and the study of their properties.
![[1860-5397-20-24-i1]](/bjoc/content/inline/1860-5397-20-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Comparison of basicity (in water scale) and synthetic availability of quinoline-type azaarenes and "proton sponge" 1.
Scheme 1: Comparison of basicity (in water scale) and synthetic availability of quinoline-type azaarenes and ...
For example, quinoquinoline 3 is synthesized in six steps from diamine 2 with a total yield not exceeding 10% [9]. This azaarene still remains a poorly available compound despite the rich chemistry of its functional derivatives, the interesting tautomeric, proton acceptor, and ligating properties. These were recently discovered and further developed by the Plieger’s group, who were also able to somewhat optimize the initial Staab’s approach [11-14]. At the same time, we showed that its acenaphthene analog 5 can be synthesized in a single step in 55–60% yield directly from diamine 4 [15]. This result is contrary to the initial report [16]. Compound 5 still has the properties of a rather strong heterocyclic base, having higher basicity than “proton sponge” (Scheme 1) [15]. Although the physicochemical properties of acenaphthene 5, including structure, protonation, and luminescence, have recently been studied by us in sufficient detail [15], nothing is known so far about the possibility of chemical modification of this molecule. It should be emphasized that the presence of a dimethylene moiety in the peri-positions of the naphthalene system will not only make molecule 5 (and derivatives) more rigid and flat when compared to compound 3 but it will also affect its reactivity and the sites of functionalization. This work is devoted to the clarification of this circumstance with substitution and elimination reactions chosen as the key transformations. The effect of functional groups on the further chemistry and basicity of the newly synthesized derivatives is also considered.
Results and Discussion
Amination, dehydrogenation, and supramolecular aggregation
Direct amination of quinoquinoline 5 could potentially lead to 2(11)-substituted amines 6 (Figure 1), the basicity of which must obviously be higher than that of the starting heterocycle 5. To this end, we conducted a series of experiments on its oxidative amination by varying the reaction conditions (time, temperature, addition of n-butyllithium to increase the nucleophilicity of amines), and reagents (amine, oxidizing agent) (for details and literature sources, see Supporting Information File 1, Table S1).
![[1860-5397-20-24-1]](/bjoc/content/figures/1860-5397-20-24-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Suggested amination products 6 and two resonance forms of dianion 7.
Figure 1: Suggested amination products 6 and two resonance forms of dianion 7.
In most experiments, only starting compound 5 was isolated almost quantitatively from the reaction mixture. Azaarene 5 proved to be resistant to the action of reagents even in the presence of KMnO4 at 100 °C. The variation of the nucleophile did not give positive results either. For example, the replacement of dimethylamine with higher-boiling piperidine and n-butylamine or using liquid ammonia, which is highly reactive in such transformations, dipyridoacenaphthene 5 still returned unchanged, although quinoline and its derivatives are easily aminated under the same conditions [17]. No interaction occurred under the conditions of the Chichibabin reaction in an attempt to aminate compound 5 with sodium amide in N,N-dimethylaniline at 140–155 °C for an hour. Thus, quinoquinoline 5, despite the presence of two pyridine nitrogen atoms, is extremely inert towards nucleophilic and oxidative amination reactions. Although molecule 5 does not explicitly contain deactivating electron-donating substituents such as alkoxy and dialkylamino (except for alkyl groups), the observed inertness may be associated with the increased basicity of the starting substrate and its structural organization (proximity of the two nitrogen atoms). The revealed inertness may also be rationalized via the ionization of the starting dipyridoacenaphthene system 5 to an anion or even dianion 7 (at least in equilibrium) under the action of an excess of nucleophiles as bases. Such dianions are characteristic of acenaphthene and have been repeatedly detected in subsequent transformations [18,19]. In our case, the CH-acidity of the CH2CH2 bridge should be even higher under the action of pyridine rings, and, if dianion 7 forms (resonance form 7b will prevail in this case, Figure 1), it will be inactive to attack by nucleophiles. The behavior of acenaphthene 5 could be clarified further using its naphthalene analog 3, which lacks benzylic CH2 protons, but there is no information in the literature about its activity/inactivity in amination reactions.
It is known that acenaphthylenes are usually readily formed from acenaphthenes by dehydrogenation with chloranil, dichlorodicyanobenzoquinone (DDQ) or active MnO2 on reflux in toluene/xylene and other inert solvents. However, attempts to obtain acenaphthylene 8 (Figure 2) as a fully conjugated analog of acenaphthene 5 by classical methods were unsuccessful (Supporting Information File 1, Table S2). In all experiments, the starting compound remained unchanged or bound into a sparingly soluble precipitate even in high-boiling solvents such as o-dichlorobenzene or nitrobenzene.
![[1860-5397-20-24-2]](/bjoc/content/figures/1860-5397-20-24-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Targeted dipyridoacenaphthylene 8.
Figure 2: Targeted dipyridoacenaphthylene 8.
For example, quinoquinoline 5 reacts with chloranil upon heating in toluene to give a dark brown, high-melting product, in which, however, the methylene bridge remains unchanged. According to 1H NMR spectroscopy and combustion analysis data, this is complex 9 with a composition close to the ratio 1:1 (Scheme 2).
![[1860-5397-20-24-i2]](/bjoc/content/inline/1860-5397-20-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of complex 9 and its slow hydrolytic degradation into protic salt 5·HCl.
Scheme 2: Formation of complex 9 and its slow hydrolytic degradation into protic salt 5·HCl.
Complex 9 is insoluble in most organic solvents, but it turned out to be unstable in DMSO-d6 solution, as evidenced by monitoring its 1Н NMR spectra (Supporting Information File 1, Figure S1). Figure S1a shows the spectrum of the starting quinoline 5, Figure S1b represents complex 9 30 min after dissolution, while Figure S1c displays complex 9 three days later. Thus, in the spectrum of complex 9, there is a distinct downfield shift of the signals of all protons in comparison with the same signals of base 5. After three days, the shift noticeably increases simultaneously with the appearance of a signal at 17.5 ppm. The latter can be uniquely attributed to the signal of the proton chelated by the aza groups of the pyridine rings. Indeed, this spectrum almost completely coincides with the spectrum of protonated quinoquinoline, as shown in Figure S1d, depicting the spectrum of picrate 5Н+PicO−, in the cationic part of which a similar intramolecular NHN hydrogen bond is realized [15]. To understand the structure of the resulting complex, we tried to grow its crystals from acetonitrile by co-evaporating solutions of quinoline 5 and chloranil at room temperature. Interestingly, in this case, hydrolytic degradation of chloranil also occurred during crystallization (base 5 could act as a catalyst for such degradation), because of which yellowish needles were obtained (neither 5 nor chloranil crystallize in this form), which turned out to be the hydrochloride dihydrate of compound 5. As the XRD study of the crystals showed (Figure 3), the molecular and crystal structure of the isolated compound is strongly dominated, on the one hand, by intra- and intermolecular hydrogen bonds with the participation of N, Cl, and O heteroatoms (forming an endless slightly corrugated ribbon), and on the other hand, by π-stacking of the antiparallel protonated dipyridoacenaphthene fragments (two-dimensional dense stacks with an interplanar distance of 3.377 Å). Their combination is the main driving force behind the formation of the final supramolecular zipper structure.
![[1860-5397-20-24-3]](/bjoc/content/figures/1860-5397-20-24-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular and crystal structure of salt 5·HCl·2H2O is strongly dominated by severe H-bonding (blue dotted lines) and π-stacking (one preorganized layer along the c-axis is shown).
Figure 3: Molecular and crystal structure of salt 5·HCl·2H2O is strongly dominated by severe H-bonding (blue ...
Interestingly, the antiparallel orientation of the closely spaced cationic fragments of base 5 can be reversed to the opposite. This can be achieved with 4,6-dichlororesorcinol, a well-known molecular organizer and coordinating agent [20,21]. Thus, the joint crystallization of dipyridoacenaphthene 5 and 4,6-dichlororesorcinol in a 2:1 ratio leads to the formation of co-crystals, in which, as judged by the X-ray data, the supramolecular organization is again in action (Figure 4). Two molecules of the base are almost parallel to each other (the distance between the π-systems of two molecular planes is 3.551 Å with the divergence angle between them of only 1.33°) and are simultaneously connected by two bifurcated hydrogen bonds with the hydroxy groups of dichlororesorcinol.
![[1860-5397-20-24-4]](/bjoc/content/figures/1860-5397-20-24-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Selected images of the supramolecular organization of two molecules of base 5 held by 4,6-dichlororesorcinol in an almost parallel manner showing side view (а), view with the resorcinol molecule directed to the viewer (b), and view above the azaarene cycles showing their almost complete overlap (с). Again, π-stacking and H-bonding (blue dotted lines) strongly dominate in this structure.
Figure 4: Selected images of the supramolecular organization of two molecules of base 5 held by 4,6-dichloror...
What would be the crystal structure of quinoquinoline 5 free of foreign particles? This is not an easy question to answer, since the superbasic nature of quinoquinolines, their planar structure, and very easy coordination to acidic and electrophilic sites (including water [15,22] or the C–H bond of chloroform [11]) almost always lead to co-crystallization. For example, there is no such crystallographic information for quinoquinoline 3 itself. In the present work, we succeeded in filling this gap by growing crystals of base 5 from pure acetonitrile. It turned out that molecule 5 is capable of self-association through multiple C–H…N–H-bond-like contacts involving pyridyl C(3)H and C(4)H protons (Figure 5). These intermolecular contacts, whose value lies in the range of 2.51–2.61 Å, strongly resemble the bifurcated hydrogen bonds so characteristic of base 5, additionally reinforced by π-stacking between the terminal components in each H-associated triad (the shortest distance between the antiparallel π-systems of two molecular planes here is 3.353 Å).
![[1860-5397-20-24-5]](/bjoc/content/figures/1860-5397-20-24-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fragment of the crystal packing of neutral dipyridoacenaphthene 5 showing self-association via multiple C–H…N contacts (blue dotted lines) between three independent molecules.
Figure 5: Fragment of the crystal packing of neutral dipyridoacenaphthene 5 showing self-association via mult...
Interestingly, as the acidity of the neighboring component in the crystal structure and the degree of proton transfer from it to the nitrogen atoms of quinoquinoline 5 decrease, the internitrogen distance regularly increases (N…N, Å): 2.709 (HCl), 2.808 (4,6-dichlororesorcinol), 2.813–2.835 (base 5). At the same time, in contrast to quinoquinoline 3, which sometimes adopts a twisted shape [11,13], molecule 5 each time remains almost flat.
Nitration, nucleophilic methoxylation, and basicity measurements
The nitration reaction of compound 5 should proceed in the same way as in other quinolines, at the benzene ring, and the resulting nitro compounds could potentially be subjected to further transformations, including nucleophilic substitution of nitro groups. Indeed, under the action of a small excess of the nitrating mixture, dipyridoacenaphthene 5 undergoes double nitration at positions 5 and 8 already at room temperature (Scheme 3). The overall yield of the main product 10 turned out to be high, but the substance contained a hard-to-separate impurity in an amount of up to 12%, to which, judged by the high-field position of the signals in the corresponding proton spectrum, was assigned the structure of the intermediate mononitro derivative 11 (Supporting Information File 1, Figure S2).
![[1860-5397-20-24-i3]](/bjoc/content/inline/1860-5397-20-24-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Dinitration of compound 5 and the initially assumed admixture 11.
Scheme 3: Dinitration of compound 5 and the initially assumed admixture 11.
Hoping to avoid formation of the “mononitro derivative” impurity, we increased the reaction time, the amount and composition of the nitrating mixture, and the temperature, but according to the results of 1H NMR spectra and TLC analysis, the second component was still present. This “permanent impurity” cannot be separated by chromatography or recrystallization, but it can be eliminated to some extent by washing the nitration products with hot chloroform, in which the impurity is slightly better soluble. This, however, is associated with losses of the main substance as well (Supporting Information File 1, Figure S3). Then, we decided to synthesize the mononitro derivative 11 purposefully by the action of one equivalent of the nitrating mixture. The reaction proceeded surprisingly well at 0 °C in several minutes with a good yield (Scheme 4). The spectral data fully confirmed the purity and asymmetric structure of product 11. It should be emphasized that the isolation and purification of nitro compounds 10 and 11 is complicated by their low solubility in traditional organic solvents, sensitivity to sunlight (especially on adsorbents), and basic dipolar solvents (DMSO, for example, causes rather rapid degradation) [23]. Note that nitro derivatives 10 or 11 are not formed when base 5 is kept in nitric acid (25 °C, excess of 65% HNO3, 24 h) which returns the starting compound unchanged.
![[1860-5397-20-24-i4]](/bjoc/content/inline/1860-5397-20-24-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
After that step, it became obvious that the second component formed in the dinitration reaction is not the mononitro derivative 11, but acenaphthylene 12 (Figure 6).
![[1860-5397-20-24-6]](/bjoc/content/figures/1860-5397-20-24-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Structure of dinitroacenaphthylene 12.
Figure 6: Structure of dinitroacenaphthylene 12.
To prove this hypothesis, as well as to compare the ease of dehydrogenation of dinitroacenaphthene 10 with respect to the initial substrate 5, it was decided to carry out its oxidation to 12 by the traditional method – the action of chloranil in boiling benzene or chloroform (in the latter, the solubility of the components is somewhat better, although the temperature of the process decreases). At the end of the synthesis, the reaction mass was treated with a potassium hydroxide solution, and the oxidation product was isolated by chromatography. Nitroacenaphthylene 13 can also be obtained similarly (Scheme 5). Thus, although nitro groups usually hinder the dehydrogenation of acenaphthenes, in our case, the opposite trend is observed. We believe that the effect of nitro groups on the success of dehydrogenation here is associated with three circumstances: 1) a decrease in the basicity of the initial heterocycle 5 and, accordingly, a decrease in the degree of base-induced degradation of chloranil, 2) difficulties in the formation of molecular complexes due to the presence of 5(8) substituents, emerging from the plane of the π-system (see the previous section), and 3) acidification of the hydrogen atoms of the CH2 groups, which can facilitate their subsequent elimination.
![[1860-5397-20-24-i5]](/bjoc/content/inline/1860-5397-20-24-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Dehydrogenation of compounds 10 and 11.
Scheme 5: Dehydrogenation of compounds 10 and 11.
The analysis of the 1Н NMR spectrum not only confirmed the structure of compound 12, but also showed its identity to the sample obtained by the action of the nitrating mixture (Supporting Information File 1, Figures S2 and S7).
Since the nitro groups in dinitroquinoquinoline 10 are formally in conjugated positions relative to the pyridine nitrogen atoms, they could potentially undergo a nucleophilic substitution. Indeed, upon boiling with an excess of sodium methoxide in methanol, the crude dinitration product 10(12) gives up to 6% of a new substance with low mobility on sorbents and blue luminescence under UV light. Its spectral analysis confirmed the symmetrical structure with two methoxy groups, however, the CH2CH2 bridge was absent and the corresponding acenaphthylene 14 was obtained instead (Scheme 6).
![[1860-5397-20-24-i6]](/bjoc/content/inline/1860-5397-20-24-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Nucleophilic methoxylation of compounds 10(12).
Scheme 6: Nucleophilic methoxylation of compounds 10(12).
Considering the low yield of dimethoxy product 14, its potential source could be dinitroacenaphthylene 12, which, as mentioned above, turned out to be a common impurity in compound 10. The possibility of a double nucleophilic substitution without dehydrogenation was tested in a separate experiment with pure dinitro compound 12 taken as a starting material. Indeed, this variant produces the same dimethoxyacenaphthylene 14 in a noticeably higher yield (Scheme 6). In this case, the participation of the acenaphthylene derivative seems quite logical, since this should facilitate the SNAr reaction and inhibit the formation of type 7 anions (under the action of methoxide as a base), which are inactive to subsequent nucleophilic attack.
Dimethoxyacenaphthylene 14 is easily protonated, and its protic salt has been fully characterized as tetrafluoroborate 14H+BF4−. The 1H NMR spectrum of this salt confirmed the symmetrical structure of the heterocyclic cation with a chelated intramolecular [NHN]+ bond, whose proton in CD3CN solution resonates at 17.22 ppm (Supporting Information File 1, Figure S13). This is a rather low value for chelate-type cations, but at the same time, it is quite logical, as molecule 14 contains a short CH=CH bridge, which increases the internitrogen distance and stretches (that is, weakens) the intramolecular hydrogen bond. For comparison, in the protonated cation of the starting diazine 5 in CD3CN, the chemical shift of the “acidic” proton is observed at 18.02 ppm [15]. Next, we evaluated the pKa value of base 14 by a competitive method in acetonitrile (NMR transprotonation involving an equivalent amount of "proton sponge" 1 as a reference compound) [6]. Additionally, we measured the basicity of unsubstituted compound 5 in acetonitrile for the first time by the same method and the results are given in Figure 7; the pKa values for compounds 1 and 3 are taken from references [24,25].
![[1860-5397-20-24-7]](/bjoc/content/figures/1860-5397-20-24-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Basicity of key compounds in acetonitrile.
Figure 7: Basicity of key compounds in acetonitrile.
As can be seen, although diazine 14 turned out to be more basic than diamine 1, the cumulative effect of all functional groups in this compound led to a slight drop in the pKa value compared to quinoquinolines 3 and 5, despite the presence of two electron-donating methoxy groups.
Bromination and tele-elimination
As preliminary experiments showed, dipyridoacenaphthene 5 is not brominated by molecular bromine in chloroform or acetic acid. The action of the NBS–DMF system, previously proposed for the electrophilic bromination of alkylaromatic compounds [26], leaves substrate 5 unchanged at room temperature, and when heated to 75 °C for several days causes its gradual degradation. Obviously, in our case, the activating effect of the CH2CH2 fragment in the naphthalene part of molecule 5 is insufficient against the background of the presence of two pyridine rings in its structure. In this regard, we turned to concentrated sulfuric acid as a reaction medium and activator of NBS, as was previously shown by the example of a very successful bromination of 6-methylquinoline at position 5 with a preparative yield of 74% [27]. Indeed, under the new conditions, we obtained dibromo derivative 15 in high yield without heating and subsequent purification (Scheme 7).
![[1860-5397-20-24-i7]](/bjoc/content/inline/1860-5397-20-24-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Electrophilic bromination of compound 5.
Scheme 7: Electrophilic bromination of compound 5.
The structure of compound 15 was confirmed by a combination of spectral methods, in particular, the disappearance of a singlet from H-5,8 protons at 7.8–7.9 ppm in the starting material 5 during functionalization (nitration, bromination), unambiguously indicates the occurrence of substituents precisely in these positions.
An attempt to dehydrogenate dibromide 15 was unsuccessful: the initial substrate remained unchanged after 2.5 hours of reflux in chloroform with one equivalent of chloranil (these conditions are practically similar to those used for the dehydrogenation of dinitro compound 10). This result brings dibromide 15 closer in chemical properties to the parent compound 5, which also cannot be converted into the corresponding acenaphthylene by direct dehydrogenation (see above). On the other hand, heating dibromide 15 with an excess of pyrrolidine for the purpose of nucleophilic substitution of the bromo-substituent led to a rather unexpected result. After cooling, dilution with water, basification, and extraction from the reaction mass, a single substance was isolated in almost quantitative yield, which turned out to be monosubstituted acenaphthylene 16, rather than the expected disubstituted acenaphthene 17 (Scheme 8).
![[1860-5397-20-24-i8]](/bjoc/content/inline/1860-5397-20-24-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: tele-Elimination upon interaction of dibromide 15 with pyrrolidine.
Scheme 8: tele-Elimination upon interaction of dibromide 15 with pyrrolidine.
In addition to the spectral data confirming the composition and asymmetric structure of compound 16, a clear sign of the emerging acenaphthylene system is its yellow-orange color, which distinguishes the UV-active (yellow-green luminescence) acenaphthylene 16 from the light-beige UV-inactive starting acenaphthene 15. It is not possible to fix any intermediate products in this unusual transformation proceeding as tele-elimination with simultaneous nucleophilic substitution. Thus, carrying out the reaction under milder conditions (130 °C, 6 h) leads only to a mixture of 15 and 16 in a 45:55 ratio.
It should be recalled here that tele-elimination refers to the cleavage of molecular fragments located further than in the vicinal positions (at least three to five carbons between the hydrogen and bromine atoms in the case of 15 to give 16). This rare transformation usually results in the formation of unsaturated products, often in the form of polyenes or alkynes [28]. The non-standard transition method found here in the acenaphthene–acenaphthylene pair is a previously unknown approach for the synthesis of acenaphthylenes based on tele-elimination (see also below).
Changing the nucleophile to methoxide we tried to obtain an analog of compound 14 with the saturated CH2–CH2 bridge. While short-time heating leaves dibromide 15 mainly unchanged, refluxing for 3 days in the MeONa/MeOH system led to the formation of new compounds alongside the dibromo derivative 15. Surprisingly, the 1H NMR spectrum showed the presence of acenaphthylene 8 and quinoline 5 as the major species in proportionate quantities. The use of sodium ethoxide in EtOH allowed us to carry out the reaction with full conversion in 2 days. Unfortunately, the admixtures and tarring formed in sufficient quantity made it difficult to purify compound 8. At the same time, isolation of the new product 8 turned out to be more convenient on using the simple KOH/EtOH system. These conditions did not affect the yield of acenaphthylene 8 (Scheme 9). Compound 8 possesses fluorescence in solutions and the solid state both as the base and in the protonated form.
![[1860-5397-20-24-i9]](/bjoc/content/inline/1860-5397-20-24-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Interaction of dibromide 15 with anionic bases.
Scheme 9: Interaction of dibromide 15 with anionic bases.
Thus, the unusual products of base treatment on dibromide 15 can be formally considered as a result of tele-elimination of bromine with the simultaneous shift of the two hydrogen atoms from the CH2CH2 bridge to positions 5(8) of the naphthalene system (formation of acenaphthylene 8) or as a result of double protodebromination (giving acenaphthene 5). Overall, the observed process resembles a redox transformation. Benzyl-type anions, which have hydride mobility and are formed in an alkaline environment from 15, may act as a reducing agent here. We tried to stop this transformation (debromination of 15 under the action of hydroxides or alkoxides) at early stages to catch possible intermediates, but without success (for example, in the 1H NMR spectrum of the mixture there are no signals of methoxy groups when using MeONa). Additional experiments show that under the reaction conditions, quinoquinolines 5 and 8 do not transform into each other (prolonged boiling in an alcoholic KOH leaves them unchanged), and, therefore, are formed independently.
Next, the molecular structures of key molecules 5 and 8 in the form of their tetrafluoroborate salts were compared. For this, both compounds 5·HBF4 and 8·HBF4 were recrystallized from acetonitrile and subjected to XRD analysis under the same conditions. Selected data obtained are shown in Table 1. As can be seen, in both protonated quinoquinoline systems, an intramolecular hydrogen bond is realized (strongly asymmetric in crystals, but dynamically symmetric in solution), however, due to the noticeably larger internitrogen distance in cation 8H+, the H bond in it is significantly weakened, as evidenced by a lower degree of deshielding of the chelated NH proton (cf. δNH values; see also data for protonated 14) and a shorter counterion–NH proton contact (Table 1). Of course, the reason for this is the appearance in molecule 8 of a short CH=CH bridge, which enhances in-plane deformations of the entire molecular system [29]. As a result, the distance between the pyridine nitrogen atoms and, at the same time, the molecular rigidity naturally increase in the series 3 → 5 → 8 (“clothespin” effect) [30].
Table 1: Comparison of selected solid-state (XRD) and solution (1H NMR) parameters of dipyridoacenaphthene 5 (left) and dipyridoacenaphthylene 8 (right) taken as the monoprotonated tetrafluoroborates (the shortest distances between the N–H proton and the counterion are also shown).
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-24-i10.png?max-width=637&scale=1.0)
|
||
| Parameter | 5·HBF4 | 8·HBF4 |
| N…N distance, Å | 2.697 | 2.712 |
| N–H distance, Å | 0.93 | 0.93 |
| N…H distance, Å | 1.94 | 1.96 |
| C9–C8 distance, Å | 1.554 | 1.354 |
| ∠NHN angle, ° | 137 | 136 |
| δNH, CD3CN | 18.02 | 17.25 |
| δH-2(11), CD3CN | 9.19 | 9.06 |
The crystal packing patterns in salts 5·HBF4 and 8·HBF4 are quite similar. The main factor here continues to be the tendency of almost flat disk-shaped heterocyclic cations to π-stacking, leading to the formation of dense columns with anions in between (Supporting Information File 1, Figure S22). Flat dipyridoacenaphthylene cations 8H+ give a denser packing, which, with an interplanar distance of only 3.328 Å, is the closest among all the studied compounds.
Conclusion
Using single crystal XRD technique, dipyridoacenaphthene tetrafluoroborate, dipyridoacenaphthene chloride dihydrate, its 2:1 complex with 4,6-dichlororesorcinol, and neutral dipyridoacenaphthene as a self-associate were obtained and structurally characterized for the first time. The dominant feature of all crystal structures is the intramolecular NHN hydrogen bonding in combination with π-stacking of almost planar diazaarene fragments, leading to pronounced supramolecular aggregation. Although dipyridoacenaphthene does not undergo nucleophilic amination and dehydrogenation under a wide range of conditions, its 5(8)-nitro derivatives can be transformed under mild conditions into the corresponding acenaphthylene by the classical method using chloranil.
The potential activity of 5(8)-nitro groups in dipyridoacenaphthylene in nucleophilic substitution reactions was shown, and a 5,8-dimethoxy derivative containing both donor substituents and an acenaphthylene fragment was synthesized. Measurement of its basicity in acetonitrile medium showed that the combined effect of two methoxy groups and the acenaphthylene fragment is negative, leading to a decrease in basicity by 0.5 pKa units compared to unsubstituted dipyridoacenaphthene, although 5,8-dimethoxydipyridoacenaphthylene is still more basic than the naphthalene "proton sponge".
A convenient and high yielding method was proposed to brominate dipyridoacenaphthene at positions 5 and 8 using a H2SO4/NBS system. The resulting dibromide turned out to be inert to dehydrogenation with chloranil, however, when heated with neutral (pyrrolidine) and anionic (NaOEt, KOH) bases, it can smoothly undergo tele-elimination, giving either functional derivatives or even unsubstituted and previously unknown dipyridoacenaphthylene. Since the discovered transformations are implemented in reasonable yields, they can be recommended as a new synthetic approach to acenaphthylene systems.
Supporting Information
CCDC 2294253 (for 2(5)·(4,6-dichlororesorcinol)), 2294254 (for 5·HBF4), 2294255 (for 8·HBF4), 2294256 (for 5), 2294257 (for 5·HCl·2H2O) contain supporting crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/data_request/cif.
| Supporting Information File 1: Additional experimental and XRD information, synthetic procedures, copies of NMR spectral data for new compounds. | ||
| Format: PDF | Size: 3.3 MB | Download |
Acknowledgements
We thank the Scientific and Educational Laboratory of Resonance Spectroscopy (Department of Natural and High Molecular Compounds Chemistry, Southern Federal University) for NMR measurements and Research Park of St. Petersburg State University for selected XRD and HRMS measurements.
Funding
This research was supported by the Russian Science Foundation (grant no. 23-23-00039, https://rscf.ru/en/project/23-23-00039/) at the Southern Federal University.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Narwal, S.; Kumar, S.; Verma, P. K. Res. Chem. Intermed. 2017, 43, 2765–2798. doi:10.1007/s11164-016-2794-2
Return to citation in text: [1] -
Lewinska, G.; Sanetra, J.; Marszalek, K. W. J. Mater. Sci.: Mater. Electron. 2021, 32, 18451–18465. doi:10.1007/s10854-021-06225-6
Return to citation in text: [1] -
Desai, N. C.; Kotadiya, G. M.; Trivedi, A. R. Bioorg. Med. Chem. Lett. 2014, 24, 3126–3130. doi:10.1016/j.bmcl.2014.05.002
Return to citation in text: [1] -
Mittal, R. K.; Aggarwal, M.; Khatana, K.; Purohit, P. Med. Chem. 2023, 19, 31–46. doi:10.2174/1573406418666220303151919
Return to citation in text: [1] -
Djurdjevic, S.; Leigh, D. A.; McNab, H.; Parsons, S.; Teobaldi, G.; Zerbetto, F. J. Am. Chem. Soc. 2007, 129, 476–477. doi:10.1021/ja067410t
Return to citation in text: [1] -
Ozeryanskii, V. A.; Pozharskii, A. F. Eur. J. Org. Chem. 2018, 3298–3305. doi:10.1002/ejoc.201800652
Return to citation in text: [1] [2] -
Peran, N.; Maksić, Z. B. Chem. Commun. 2011, 47, 1327–1329. doi:10.1039/c0cc03386c
Return to citation in text: [1] -
Pozharskii, A. F.; Ozeryanskii, V. A.; Filatova, E. A. Chem. Heterocycl. Compd. 2012, 48, 200–219. doi:10.1007/s10593-012-0983-5
Return to citation in text: [1] [2] -
Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1987, 26, 460–461. doi:10.1002/anie.198704601
Return to citation in text: [1] [2] [3] -
Staab, H. A.; Diehm, M.; Krieger, C. Tetrahedron Lett. 1994, 35, 8357–8360. doi:10.1016/s0040-4039(00)74406-6
Return to citation in text: [1] -
Shaffer, K. J.; McLean, T. M.; Waterland, M. R.; Wenzel, M.; Plieger, P. G. Inorg. Chim. Acta 2012, 380, 278–283. doi:10.1016/j.ica.2011.09.046
Return to citation in text: [1] [2] [3] -
Shaffer, K. J.; Parr, D. C.; Wenzel, M.; Rowlands, G. J.; Plieger, P. G. Eur. J. Org. Chem. 2012, 6967–6975. doi:10.1002/ejoc.201201131
Return to citation in text: [1] -
Rowlands, G. J.; Severinsen, R. J.; Buchanan, J. K.; Shaffer, K. J.; Jameson, H. T.; Thennakoon, N.; Leito, I.; Lõkov, M.; Kütt, A.; Vianello, R.; Despotović, I.; Radić, N.; Plieger, P. G. J. Org. Chem. 2020, 85, 11297–11308. doi:10.1021/acs.joc.0c01428
Return to citation in text: [1] [2] -
Buchanan, J. K.; Severinsen, R. J.; Buchner, M. R.; Thomas-Hargreaves, L. R.; Spang, N.; John, K. D.; Plieger, P. G. Dalton Trans. 2021, 50, 16950–16953. doi:10.1039/d1dt03367k
Return to citation in text: [1] -
Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Buu-Hoï, N. P.; Dufour, M.; Jacquignon, P. J. Chem. Soc. C 1968, 2070–2072. doi:10.1039/j39680002070
Return to citation in text: [1] -
Tondys, H.; van der Plas, H. C.; Woźniak, M. J. Heterocycl. Chem. 1985, 22, 353–355. doi:10.1002/jhet.5570220226
Return to citation in text: [1] -
Kershner, L. D.; Gaidis, J. M.; Freedman, H. H. J. Am. Chem. Soc. 1972, 94, 985–986. doi:10.1021/ja00758a044
Return to citation in text: [1] -
Van Loo, M. E.; Lugtenburg, J.; Cornelisse, J. Eur. J. Org. Chem. 1998, 1907–1914. doi:10.1002/(sici)1099-0690(199809)1998:9<1907::aid-ejoc1907>3.0.co;2-s
Return to citation in text: [1] -
Papaefstathiou, G. S.; MacGillivray, L. R. Org. Lett. 2001, 3, 3835–3838. doi:10.1021/ol0165948
Return to citation in text: [1] -
Grobelny, A. L.; Rath, N. P.; Groeneman, R. H. CrystEngComm 2018, 20, 3951–3954. doi:10.1039/c8ce00447a
Return to citation in text: [1] -
Krieger, C.; Newsom, I.; Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1989, 28, 84–86. doi:10.1002/anie.198900841
Return to citation in text: [1] -
van den Braken‐van Leersum, A. M.; Spijker, N. M.; Lugtenburg, J.; Cornelisse, J. Recl. Trav. Chim. Pays-Bas 1987, 106, 628–640. doi:10.1002/recl.19871061205
Return to citation in text: [1] -
Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I. A. J. Org. Chem. 2005, 70, 1019–1028. doi:10.1021/jo048252w
Return to citation in text: [1] -
Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P. G.; Vianello, R.; Leito, I. Eur. J. Org. Chem. 2017, 4475–4489. doi:10.1002/ejoc.201700749
Return to citation in text: [1] -
Mitchell, R. H.; Lai, Y.-H.; Williams, R. V. J. Org. Chem. 1979, 44, 4733–4735. doi:10.1021/jo00393a066
Return to citation in text: [1] -
Tochilkin, A. I.; Kovel'man, I. R.; Prokof'ev, E. P.; Gracheva, I. N.; Levinskii, M. V. Chem. Heterocycl. Compd. 1988, 24, 892–897. doi:10.1007/bf00479345
Return to citation in text: [1] -
Brandsma, L. Acetylenes, Allenes and Cumulenes by Elimination Reactions. Synthesis of Acetylenes, Allenes and Cumulenes; Academic Press: Amsterdam, Netherlands, 2004; pp 203–228. doi:10.1016/b978-012125751-4/50011-4
Return to citation in text: [1] -
Pozharskii, A. F.; Ozeryanskii, V. A.; Starikova, Z. A. J. Chem. Soc., Perkin Trans. 2 2002, 318–322. doi:10.1039/b106725g
Return to citation in text: [1] -
Filatova, E. A.; Ermolenko, E. A.; Pozharskii, A. F.; Ozeryanskii, V. A.; Demidov, O. P.; Chernyshev, A. V.; Metelitsa, A. V.; Gulevskaya, A. V. Org. Biomol. Chem. 2023, 21, 3388–3401. doi:10.1039/d3ob00286a
Return to citation in text: [1]
| 29. | Pozharskii, A. F.; Ozeryanskii, V. A.; Starikova, Z. A. J. Chem. Soc., Perkin Trans. 2 2002, 318–322. doi:10.1039/b106725g |
| 30. | Filatova, E. A.; Ermolenko, E. A.; Pozharskii, A. F.; Ozeryanskii, V. A.; Demidov, O. P.; Chernyshev, A. V.; Metelitsa, A. V.; Gulevskaya, A. V. Org. Biomol. Chem. 2023, 21, 3388–3401. doi:10.1039/d3ob00286a |
| 1. | Narwal, S.; Kumar, S.; Verma, P. K. Res. Chem. Intermed. 2017, 43, 2765–2798. doi:10.1007/s11164-016-2794-2 |
| 2. | Lewinska, G.; Sanetra, J.; Marszalek, K. W. J. Mater. Sci.: Mater. Electron. 2021, 32, 18451–18465. doi:10.1007/s10854-021-06225-6 |
| 8. | Pozharskii, A. F.; Ozeryanskii, V. A.; Filatova, E. A. Chem. Heterocycl. Compd. 2012, 48, 200–219. doi:10.1007/s10593-012-0983-5 |
| 18. | Kershner, L. D.; Gaidis, J. M.; Freedman, H. H. J. Am. Chem. Soc. 1972, 94, 985–986. doi:10.1021/ja00758a044 |
| 19. | Van Loo, M. E.; Lugtenburg, J.; Cornelisse, J. Eur. J. Org. Chem. 1998, 1907–1914. doi:10.1002/(sici)1099-0690(199809)1998:9<1907::aid-ejoc1907>3.0.co;2-s |
| 7. | Peran, N.; Maksić, Z. B. Chem. Commun. 2011, 47, 1327–1329. doi:10.1039/c0cc03386c |
| 8. | Pozharskii, A. F.; Ozeryanskii, V. A.; Filatova, E. A. Chem. Heterocycl. Compd. 2012, 48, 200–219. doi:10.1007/s10593-012-0983-5 |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 5. | Djurdjevic, S.; Leigh, D. A.; McNab, H.; Parsons, S.; Teobaldi, G.; Zerbetto, F. J. Am. Chem. Soc. 2007, 129, 476–477. doi:10.1021/ja067410t |
| 6. | Ozeryanskii, V. A.; Pozharskii, A. F. Eur. J. Org. Chem. 2018, 3298–3305. doi:10.1002/ejoc.201800652 |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 3. | Desai, N. C.; Kotadiya, G. M.; Trivedi, A. R. Bioorg. Med. Chem. Lett. 2014, 24, 3126–3130. doi:10.1016/j.bmcl.2014.05.002 |
| 4. | Mittal, R. K.; Aggarwal, M.; Khatana, K.; Purohit, P. Med. Chem. 2023, 19, 31–46. doi:10.2174/1573406418666220303151919 |
| 17. | Tondys, H.; van der Plas, H. C.; Woźniak, M. J. Heterocycl. Chem. 1985, 22, 353–355. doi:10.1002/jhet.5570220226 |
| 11. | Shaffer, K. J.; McLean, T. M.; Waterland, M. R.; Wenzel, M.; Plieger, P. G. Inorg. Chim. Acta 2012, 380, 278–283. doi:10.1016/j.ica.2011.09.046 |
| 12. | Shaffer, K. J.; Parr, D. C.; Wenzel, M.; Rowlands, G. J.; Plieger, P. G. Eur. J. Org. Chem. 2012, 6967–6975. doi:10.1002/ejoc.201201131 |
| 13. | Rowlands, G. J.; Severinsen, R. J.; Buchanan, J. K.; Shaffer, K. J.; Jameson, H. T.; Thennakoon, N.; Leito, I.; Lõkov, M.; Kütt, A.; Vianello, R.; Despotović, I.; Radić, N.; Plieger, P. G. J. Org. Chem. 2020, 85, 11297–11308. doi:10.1021/acs.joc.0c01428 |
| 14. | Buchanan, J. K.; Severinsen, R. J.; Buchner, M. R.; Thomas-Hargreaves, L. R.; Spang, N.; John, K. D.; Plieger, P. G. Dalton Trans. 2021, 50, 16950–16953. doi:10.1039/d1dt03367k |
| 16. | Buu-Hoï, N. P.; Dufour, M.; Jacquignon, P. J. Chem. Soc. C 1968, 2070–2072. doi:10.1039/j39680002070 |
| 9. | Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1987, 26, 460–461. doi:10.1002/anie.198704601 |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 9. | Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1987, 26, 460–461. doi:10.1002/anie.198704601 |
| 10. | Staab, H. A.; Diehm, M.; Krieger, C. Tetrahedron Lett. 1994, 35, 8357–8360. doi:10.1016/s0040-4039(00)74406-6 |
| 9. | Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1987, 26, 460–461. doi:10.1002/anie.198704601 |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 11. | Shaffer, K. J.; McLean, T. M.; Waterland, M. R.; Wenzel, M.; Plieger, P. G. Inorg. Chim. Acta 2012, 380, 278–283. doi:10.1016/j.ica.2011.09.046 |
| 20. | Papaefstathiou, G. S.; MacGillivray, L. R. Org. Lett. 2001, 3, 3835–3838. doi:10.1021/ol0165948 |
| 21. | Grobelny, A. L.; Rath, N. P.; Groeneman, R. H. CrystEngComm 2018, 20, 3951–3954. doi:10.1039/c8ce00447a |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 22. | Krieger, C.; Newsom, I.; Zirnstein, M. A.; Staab, H. A. Angew. Chem., Int. Ed. Engl. 1989, 28, 84–86. doi:10.1002/anie.198900841 |
| 27. | Tochilkin, A. I.; Kovel'man, I. R.; Prokof'ev, E. P.; Gracheva, I. N.; Levinskii, M. V. Chem. Heterocycl. Compd. 1988, 24, 892–897. doi:10.1007/bf00479345 |
| 28. | Brandsma, L. Acetylenes, Allenes and Cumulenes by Elimination Reactions. Synthesis of Acetylenes, Allenes and Cumulenes; Academic Press: Amsterdam, Netherlands, 2004; pp 203–228. doi:10.1016/b978-012125751-4/50011-4 |
| 24. | Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I. A. J. Org. Chem. 2005, 70, 1019–1028. doi:10.1021/jo048252w |
| 25. | Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P. G.; Vianello, R.; Leito, I. Eur. J. Org. Chem. 2017, 4475–4489. doi:10.1002/ejoc.201700749 |
| 26. | Mitchell, R. H.; Lai, Y.-H.; Williams, R. V. J. Org. Chem. 1979, 44, 4733–4735. doi:10.1021/jo00393a066 |
| 15. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Chernyshev, A. V.; Metelitsa, A. V.; Antonov, A. S. Org. Biomol. Chem. 2019, 17, 8221–8233. doi:10.1039/c9ob01391a |
| 6. | Ozeryanskii, V. A.; Pozharskii, A. F. Eur. J. Org. Chem. 2018, 3298–3305. doi:10.1002/ejoc.201800652 |
| 11. | Shaffer, K. J.; McLean, T. M.; Waterland, M. R.; Wenzel, M.; Plieger, P. G. Inorg. Chim. Acta 2012, 380, 278–283. doi:10.1016/j.ica.2011.09.046 |
| 13. | Rowlands, G. J.; Severinsen, R. J.; Buchanan, J. K.; Shaffer, K. J.; Jameson, H. T.; Thennakoon, N.; Leito, I.; Lõkov, M.; Kütt, A.; Vianello, R.; Despotović, I.; Radić, N.; Plieger, P. G. J. Org. Chem. 2020, 85, 11297–11308. doi:10.1021/acs.joc.0c01428 |
| 23. | van den Braken‐van Leersum, A. M.; Spijker, N. M.; Lugtenburg, J.; Cornelisse, J. Recl. Trav. Chim. Pays-Bas 1987, 106, 628–640. doi:10.1002/recl.19871061205 |
© 2024 Kolupaeva et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.