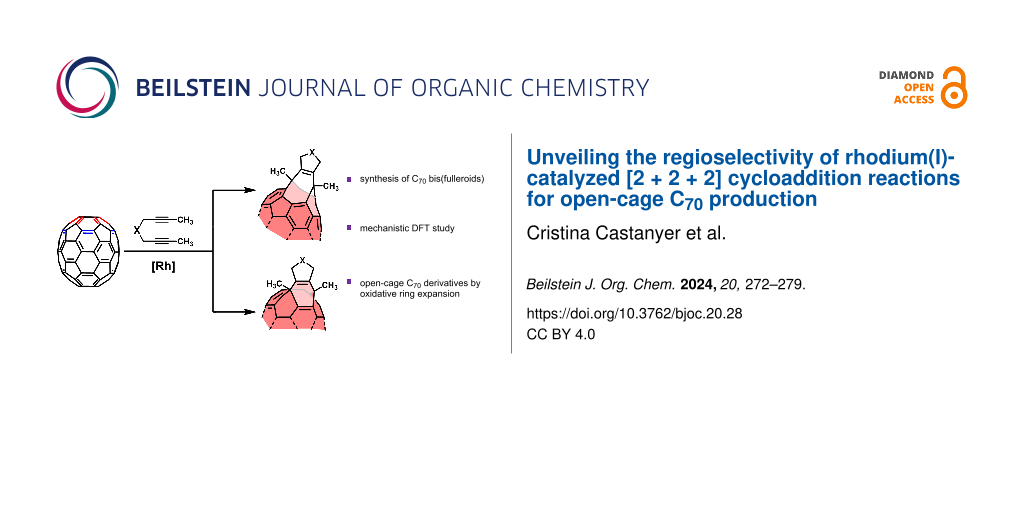
Abstract
The regioselective functionalization of fullerenes holds significant promise for applications in the fields of medicinal chemistry, materials science, and photovoltaics. In this study, we investigate the regioselectivity of the rhodium(I)-catalyzed [2 + 2 + 2] cycloaddition reactions between diynes and C70 as a novel procedure for generating C70 bis(fulleroid) derivatives. The aim is to shed light on the regioselectivity of the process through both experimental and computational approaches. In addition, the photooxidation of one of the C–C double bonds in the synthesized bis(fulleroids) affords open-cage C70 derivatives having a 12-membered ring opening.

Graphical Abstract
Introduction
The discovery of C60 (buckminsterfullerene) in 1985 [1] initiated the search for possible technological applications of fullerenes. Nowadays, applications for these carbon-based molecules have been proposed in different fields such as medicinal chemistry [2-6], materials science [7,8], energy production, storage, and delivery [9-13], and electronics and optoelectronics [14-16]. Despite fullerenes having immense promise in all of these areas, their practical applications are still in various stages of research and development.
The functionalization of fullerenes makes them versatile materials, broadening the range of potential applications [17,18]. It allows the properties of these carbon cages to be tuned, making them more soluble (especially in water for medical applications) and improving their stability, among other desirable properties. The most common reactions used to functionalize fullerenes are Diels–Alder and 1,3-dipolar cycloadditions and Bingel–Hirsch cyclopropanations [19,20].
In most cases, functionalization occurs while preserving the carbon cages. However, in other cases, some of the bonds between the C atoms of the cage are broken and the cage is opened. The first example of an open-cage fullerene was reported in 1995 by Hummelen, Prato, and Wudl [21] through the reaction of C60 with azides followed by photooxygenation. Since then, many open-cage C60 derivatives have been reported. These open-cage fullerenes can act as molecular containers. Of special interest is the procedure called molecular surgery designed by Murata et al. [22-25] in which a hole in the fullerene is opened, an atom or small molecule is introduced and then the hole is closed restoring the original cage. Among the species that have been incarcerated with this procedure, we can find He, Ne, Ar, Kr, H2, N2, O2, HF, CO, CO2, H2O, H2O2, CH4, NH3, HCOH, HCCH, and CH3OH [26,27]. The encapsulation of atoms or small molecules inside the fullerene has been found to be able to produce meaningful changes in the reactivity of the cage [28-32].
In 2018, our group reported a catalytic process to transform C60 in bis(fulleroid) derivatives [33-35]. This transformation encompassed a partially intermolecular Rh-catalyzed [2 + 2 + 2] cycloaddition reaction between diynes and C60, followed by a cage-opening through a Rh‐catalyzed di‐π‐methane rearrangement (Scheme 1). It is well-known that [6,6]-bonds (the bonds at the junction between two six-membered rings, Figure 1, left) are more reactive than [5,6]-bonds in C60 [36-38], and, not unexpectedly, the [6,6]-bond in C60 was the one involved in this [2 + 2 + 2] cycloaddition.
![[1860-5397-20-28-i1]](/bjoc/content/inline/1860-5397-20-28-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Rhodium(I)-catalyzed cycloaddition of C60 with diynes to afford bis(fulleroid) derivatives [33].
Scheme 1: Rhodium(I)-catalyzed cycloaddition of C60 with diynes to afford bis(fulleroid) derivatives [33].
![[1860-5397-20-28-1]](/bjoc/content/figures/1860-5397-20-28-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Types of [6,6]-bonds together with the [5,6]-bond of C60 with their C–C distances in pristine C60 and C70.
Figure 1: Types of [6,6]-bonds together with the [5,6]-bond of C60 with their C–C distances in pristine C60 a...
Although there are several papers reporting the opening of a hole in C70 [39-45], this chemistry has been less explored than in C60. The lower D5h symmetry of C70 compared to the Ih of C60 increases the number of possible regioisomers. Indeed, C70 has eight different bonds, half of which are different types of [6,6]-bonds, namely the α-, β-, γ-, and δ-bonds (Figure 1, right) [46]. The α- and β-bonds of C70 are the most reactive ones [47].
With this in mind, the main goal of the present work is to explore, both experimentally and computationally, the Rh-catalyzed intermolecular [2 + 2 + 2] cycloaddition reaction between diynes and C70 as a new procedure to generate C70 bis(fulleroids). We are particularly interested in the analysis of the regioselectivity of this [2 + 2 + 2] cycloaddition.
Results and Discussion
We started our study by testing the cycloaddition of N-tosyl-tethered bisalkyne 1a and C70 (Scheme 2) using our previously optimized reaction conditions for the C60 derivative [33]: that is, using 10 mol % of a mixture of [Rh(cod)2]BF4 and Tol-BINAP in o-dichlorobenzene (o-DCB) and heating at 90 °C for 4 hours. The crude reaction mass obtained with these conditions was then purified by column chromatography (toluene). After eluting unreacted pristine C70, a dark reddish fraction was isolated and analyzed by HPLC. A major peak was observed at a retention time of 17.5 minutes, which we analyzed by UV–vis spectroscopy. This peak was assigned as a bis(fulleroid) compound by comparing the spectra with the UV–vis absorption pattern exhibited by previously characterized C70 bis(fulleroids) reported by Murata et al. [43,48]. In addition, a minor peak at a retention time of 20 minutes was also observed in the HPLC chromatogram, whose UV–vis has a pattern that is similar to a previously reported α-adduct [49]. We reasoned that this minor compound was the cyclohexadiene-fused C70 intermediate, analogous to cyclohexadiene-fused C60 I (see Scheme 1), which had not completely evolved into the corresponding bis(fulleroid) product after 4 h of reaction (Figure S1 in Supporting Information File 1). Importantly, the observation of this intermediate represents an experimental proof of the proposed reaction mechanism. Confirmation that only one unit of 1a reacted with C70 in the reaction was obtained from HRMS, which gave a single peak at m/z 1138.0868 corresponding to [2a + Na]+. Further optimization was then carried out to obtain the bis(fulleroid) derivative alone (Table S1 in Supporting Information File 1). On increasing the reaction temperature to 120 °C and 180 °C the results were found to be the same, showing that 90 °C is sufficient for the reaction to proceed. In contrast, on extending the reaction time to 24 hours, the minor peak in the HPLC disappeared and only the peak corresponding to the bis(fulleroid) remained. The yield of derivative 2a was 45%. Other experiments were run using other solvents such as toluene and chlorobenzene, increasing the C70 concentration from 1.2 M to 2.4 M, and decreasing the catalytic load to 5 mol % (Table S1 in Supporting Information File 1). However, none of these trials improved the yield of bis(fulleroid) 2a.
![[1860-5397-20-28-i2]](/bjoc/content/inline/1860-5397-20-28-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rhodium-catalyzed cycloaddition of C70 with diynes 1a and 1b.
Scheme 2: Rhodium-catalyzed cycloaddition of C70 with diynes 1a and 1b.
The same reaction was run starting with malonate-tethered diyne 1b. In this case, the reaction was finished after 4 hours and bis(fulleroid) 2b was obtained with a 34% yield (Scheme 2). The corresponding compound 2b was analyzed by HPLC, giving only one peak. UV–vis experiments revealed the formation of a bis(fulleroid) derivative (Figure S2 in Supporting Information File 1). The lower yield of 2b compared to 2a is probably due to the [2 + 2 + 2] homocoupling cycloaddition of the corresponding starting diyne, which is more favorable when the tether is a malonate rather than an NTs-sulfonamide.
Among the four different [6,6]-bonds (α, β, γ, and δ) in pristine C70, α and β junctions are pyracylenic bonds, which happen to be the most reactive due to their higher degree of pyramidalization. Between both the α- and β-bonds, the higher curvature strain in α-bonds compared to β-double bonds makes the first one more reactive, leading to β-site isomers as minor products. Taking this into account, we carefully analyzed the NMR spectra of compound 2a. Analysis of 1H NMR spectra of 2a provided valuable information that confirmed the generation of two regioisomers in a 71:29 ratio (Figure 2). Comparable proportions of reactions at α- and β-bonds were systematically observed at different temperatures.
![[1860-5397-20-28-2]](/bjoc/content/figures/1860-5397-20-28-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR (CS2/CDCl3, 400 MHz) spectrum of compound 2a as a mixture of two isomers.
Figure 2: 1H NMR (CS2/CDCl3, 400 MHz) spectrum of compound 2a as a mixture of two isomers.
The protons of the two methyl groups around δ 2.30 ppm were used as diagnostic signals. For the major isomer (red dots, Figure 2), the spectrum exhibits two singlets at δ = 2.27 and 2.28 ppm, corresponding to the two different methyl groups in the six-membered ring formed in the cycloaddition and coming from the starting diyne 1a (highlighted in red in Scheme 2). In contrast, for the minor isomer (green dots), which has Cs symmetry, the two methyl groups (highlighted in green in Scheme 2) appear as a single peak at δ = 2.40 ppm. A second signal that also helps us to determine the ratio between the two isomers is the methyl of the tosyl group. For the major isomer the methyl appears at δ = 3.24 ppm, whereas for the minor isomer the peak is observed at δ = 2.62 ppm. Considering that isomer α has no symmetry and isomer β has Cs symmetry [50], we can conclude that the major product formed was the α-isomer, as previously anticipated. All attempts to separate the two isomers by column chromatography and preparative TLC were unsuccessful. Malonate-tethered compound 2b had the same spectroscopic behavior as 2a though in this case the ratio in favor of the α-isomer was higher (80:20, Figure S6 in Supporting Information File 1).
To gain theoretical insight into the regioselectivity of the reaction, a density functional theory (DFT) investigation was carried out, as depicted in Figure 3. In the computations, the tosyl group was substituted by a mesyl substituent and BIPHEP was used as a model phosphine ligand instead of Tol-BINAP to reduce the computational cost. The calculations, conducted at the B3LYP-D3/cc-pVTZ-PP(SMD=o-DCB)//B3LYP-D3/cc-pVDZ-PP level (see full computational details in Supporting Information File 1), unveiled the following reaction mechanism: initially, an oxidative coupling of the two alkyne moieties of our model 1a leads to the formation of INT 1, as previously reported [33]. This step, with a Gibbs energy barrier of 25.7 kcal·mol−1, is the rate-determining step for this process. Next, INT 1 readily coordinates with a C70 molecule to generate INT 2, with this step being exergonic by 16.7 kcal·mol−1. From INT 2, the reaction can follow two distinct pathways, culminating in either an α-adduct or a β-adduct. In the α-adduct pathway (black line), a formal [2 + 2] cycloaddition occurs between the rhodacyclopentadiene moiety and a [6,6]-α-bond of C70, yielding rhodabicyclo[3.2.0]heptadiene intermediate α-INT 3. This step has a cost of 9.5 kcal·mol−1. Alternatively, a [6,6]-β-bond of C70 can be involved in this step (grey line) to produce β-INT 3, albeit with a slightly higher Gibbs energy barrier (ΔΔG = 0.9 kcal·mol−1). The formation of intermediate α-INT 3 and β-INT 3 was found endergonic by 9.3 and 7.6 kcal·mol−1, respectively. Subsequently, both site isomers of INT 3 can undergo reductive elimination with barriers of 6.9 and 9.4 kcal·mol−1 to deliver the corresponding cyclohexadiene-fused adducts, denoted as α-INT 4 and β-INT 4, which will ultimately evolve into the final bis(fulleroid) reaction products [33]. As the site-selectivity of the reaction depends on these two consecutive steps, it indicates a preference for the α-bonds over the β-bonds, consistent with the experimental findings discussed earlier. Once INT 1 is formed, for the rest of the process, the TOF determining transition state (TDTS) of the process is α/β-TS 2 and the TOF determining intermediate (TDI) is INT 2 and the energetic span (δG) is 16.2 kcal·mol−1 for the α-attack and 17.0 kcal·mol−1 for the β-attack [51,52].
![[1860-5397-20-28-3]](/bjoc/content/figures/1860-5397-20-28-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: B3LYP-D3/cc-pVTZ-PP(SMD=o-DCB)//B3LYP-D3/CC-pVDZ-PP Gibbs energy profile of the [2 + 2 + 2] cycloaddition between our model diyne 1a and C70. Comparison between α- and β-reaction pathways. Black line: α-pathway. Grey line: β-pathway. Molecular structures correspond to the α-pathway. [Rh] = [Rh(BIPHEP)]+.
Figure 3: B3LYP-D3/cc-pVTZ-PP(SMD=o-DCB)//B3LYP-D3/CC-pVDZ-PP Gibbs energy profile of the [2 + 2 + 2] cycload...
As previously described for our analogous C60 bis(fulleroids) [33], one of the double bonds of the eight-membered ring in 2a can undergo oxidative cleavage affording open-cage C70 fullerenes that bear a twelve-membered orifice. There has been considerable interest in the construction of larger orifices in C70 derivatives given that the larger cavity compared to its C60 counterpart can facilitate the encapsulation of multiple atoms and molecules [40,43,53,54]. To fulfil this objective, compound 2a was subjected to oxidative cleavage by exposing it to light in the presence of air (Scheme 3).
![[1860-5397-20-28-i3]](/bjoc/content/inline/1860-5397-20-28-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Oxidative cleavage of bis(fulleroid) derivatives 2a and 2b.
Scheme 3: Oxidative cleavage of bis(fulleroid) derivatives 2a and 2b.
Given the lower symmetry of C70 in comparison to C60, the oxidative opening of the eight-membered ring of the mixture of α- and β-isomers 2 can result in more than two oxidized isomers, depending on which double bond is cleaved. After 5 hours of irradiation, the crude mixture was purified by column chromatography, giving an inseparable mixture of different isomers. The oxygenation process was confirmed by HRMS, which gave a single peak at m/z = 1170.0756 corresponding to [3a + Na]+. On analyzing carefully the mixture by 1H NMR spectroscopy, we observed three different sets of three methyl groups corresponding to the two methyls derived from bisalkyne 1a and the methyl in the tosyl group in the spectrum (Figure 4). These results indicate that there are three regioisomers found in a ratio of 56:29:15. Two of them result from the oxidation of α-2a, whose lack of symmetry results in two different bonds available for oxidative cleavage. Site-isomer β-2a displays Cs symmetry, and thus both bonds available for oxygenation are enantiotopic. Considering that starting 2a consisted of a 71:29 mixture of α- and β-2 isomers, we assumed that α- and α’-isomers (56% + 15% = 71%) correspond to the protons marked in red and the ones marked in green might be those of the β-isomer (29%). Unfortunately, NMR experiments did not allow to differentiate between α- and β-3a derivatives. The reaction was carried out also with bis(fulleroid) derivative 2b, exhibiting the same behavior.
![[1860-5397-20-28-4]](/bjoc/content/figures/1860-5397-20-28-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR (CS2/CDCl3, 400 MHz) spectrum of compound 3a as a mixture of three isomers. X = residual toluene.
Figure 4: 1H NMR (CS2/CDCl3, 400 MHz) spectrum of compound 3a as a mixture of three isomers. X = residual tol...
Conclusion
In this study, we have explored the regioselectivity of the rhodium(I)-catalyzed [2 + 2 + 2] cycloaddition reaction between two different diynes and C70 with the objective of producing C70 bis(fulleroids). Mixtures of α- and β-site isomers were obtained, with the α-adduct being the major product of the reaction in both cases. This preference was rationalized by means of DFT calculations. Moreover, the photooxidation of one of the C–C double bonds of the new bis(fulleroids) afford open-cage C70 derivatives having a 12-membered ring opening. It is noteworthy to mention that examples of open-cage C70 derivatives are relatively scarce, likely owing to the challenges associated with their synthesis and the characterization of asymmetric structures. The findings of this study contribute to the ongoing efforts in the field of fullerene chemistry and provide a foundation for further exploration of regioselective [2 + 2 + 2] cycloaddition reactions as a means to tailor the properties of fullerenes for specific applications.
Supporting Information
| Supporting Information File 1: General materials and methods, experimental procedures and characterization of all new compounds. | ||
| Format: PDF | Size: 2.7 MB | Download |
Acknowledgements
Dr. Carles Fuertes-Espinosa is acknowledged for assistance in HPLC and UV–vis experiments.
Funding
We are grateful for the financial support from the Ministerio de Ciencia e Innovación and EU (Project PID2020-113711GB-I00 MCIN/AEI/10.13039/50110001103, FPI predoctoral grant to C.C. and Margarita Salas grant (NextGenerationEU) to A.A.) and the Generalitat de Catalunya (Project 2021-SGR-623).
Data Availability Statement
All experimental data available in published article and/or supplementary material. All computational data are available through the ioChem-BD repository: https://www.iochem-bd.org/handle/10/356060, https://doi.org/10.19061/iochem-bd-4-67.
References
-
Kroto, H. W.; Heath, J. R.; O’Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0
Return to citation in text: [1] -
Bakry, R.; Vallant, R. M.; Najam-ul-Haq, M.; Rainer, M.; Szabo, Z.; Huck, C. W.; Bonn, G. K. Int. J. Nanomed. 2007, 2, 639–649.
Return to citation in text: [1] -
Castro, E.; Hernandez Garcia, A.; Zavala, G.; Echegoyen, L. J. Mater. Chem. B 2017, 5, 6523–6535. doi:10.1039/c7tb00855d
Return to citation in text: [1] -
Panwar, N.; Soehartono, A. M.; Chan, K. K.; Zeng, S.; Xu, G.; Qu, J.; Coquet, P.; Yong, K.-T.; Chen, X. Chem. Rev. 2019, 119, 9559–9656. doi:10.1021/acs.chemrev.9b00099
Return to citation in text: [1] -
Ramos-Soriano, J.; Reina, J. J.; Illescas, B. M.; de la Cruz, N.; Rodríguez-Pérez, L.; Lasala, F.; Rojo, J.; Delgado, R.; Martín, N. J. Am. Chem. Soc. 2019, 141, 15403–15412. doi:10.1021/jacs.9b08003
Return to citation in text: [1] -
Cataldo, F.; Da Ros, T., Eds. Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes; Carbon Materials: Chemistry and Physics; Springer Netherlands: Dordrecht, Netherlands, 2008. doi:10.1007/978-1-4020-6845-4
Return to citation in text: [1] -
Montellano López, A.; Mateo-Alonso, A.; Prato, M. J. Mater. Chem. 2011, 21, 1305–1318. doi:10.1039/c0jm02386h
Return to citation in text: [1] -
Canevet, D.; Pérez, E. M.; Martín, N. Angew. Chem., Int. Ed. 2011, 50, 9248–9259. doi:10.1002/anie.201101297
Return to citation in text: [1] -
Kim, Y.; Cook, S.; Tuladhar, S. M.; Choulis, S. A.; Nelson, J.; Durrant, J. R.; Bradley, D. D. C.; Giles, M.; McCulloch, I.; Ha, C.-S.; Ree, M. Nat. Mater. 2006, 5, 197–203. doi:10.1038/nmat1574
Return to citation in text: [1] -
Collavini, S.; Delgado, J. L. Sustainable Energy Fuels 2018, 2, 2480–2493. doi:10.1039/c8se00254a
Return to citation in text: [1] -
Jia, L.; Chen, M.; Yang, S. Mater. Chem. Front. 2020, 4, 2256–2282. doi:10.1039/d0qm00295j
Return to citation in text: [1] -
Gopal, J.; Muthu, M.; Sivanesan, I. Polymers (Basel, Switz.) 2023, 15, 701. doi:10.3390/polym15030701
Return to citation in text: [1] -
Kausar, A. Polym.-Plast. Technol. Mater. 2023, 62, 618–631. doi:10.1080/25740881.2022.2121223
Return to citation in text: [1] -
Jariwala, D.; Sangwan, V. K.; Lauhon, L. J.; Marks, T. J.; Hersam, M. C. Chem. Soc. Rev. 2013, 42, 2824–2860. doi:10.1039/c2cs35335k
Return to citation in text: [1] -
Babu, S. S.; Möhwald, H.; Nakanishi, T. Chem. Soc. Rev. 2010, 39, 4021–4035. doi:10.1039/c000680g
Return to citation in text: [1] -
Scarselli, M.; Castrucci, P.; De Crescenzi, M. J. Phys.: Condens. Matter 2012, 24, 313202. doi:10.1088/0953-8984/24/31/313202
Return to citation in text: [1] -
Ai, M.; Chen, M.; Yang, S. Chin. J. Chem. 2023, 41, 2337–2353. doi:10.1002/cjoc.202300105
Return to citation in text: [1] -
Paukov, M.; Kramberger, C.; Begichev, I.; Kharlamova, M.; Burdanova, M. Materials 2023, 16, 1276. doi:10.3390/ma16031276
Return to citation in text: [1] -
Hirsch, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 1138–1141. doi:10.1002/anie.199311381
Return to citation in text: [1] -
Taylor, R. C. R. Chim. 2006, 9, 982–1000. doi:10.1016/j.crci.2006.01.004
Return to citation in text: [1] -
Hummelen, J. C.; Prato, M.; Wudl, F. J. Am. Chem. Soc. 1995, 117, 7003–7004. doi:10.1021/ja00131a024
Return to citation in text: [1] -
Komatsu, K.; Murata, M.; Murata, Y. Science 2005, 307, 238–240. doi:10.1126/science.1106185
Return to citation in text: [1] -
Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2008, 6083–6094. doi:10.1039/b811738a
Return to citation in text: [1] -
Hashikawa, Y.; Yasui, H.; Kurotobi, K.; Murata, Y. Mater. Chem. Front. 2018, 2, 206–213. doi:10.1039/c7qm00449d
Return to citation in text: [1] -
Hashikawa, Y.; Murata, Y. Bull. Chem. Soc. Jpn. 2023, 96, 943–967. doi:10.1246/bcsj.20230135
Return to citation in text: [1] -
Stanisky, C. M.; Cross, R. J.; Saunders, M. J. Am. Chem. Soc. 2009, 131, 3392–3395. doi:10.1021/ja809831a
Return to citation in text: [1] -
Gao, R.; Liu, Z.; Liu, Z.; Liang, T.; Su, J.; Gan, L. Angew. Chem., Int. Ed. 2023, 62, e202300151. doi:10.1002/anie.202300151
and references cited therein.
Return to citation in text: [1] -
Hashikawa, Y.; Murata, Y. Chem. – Eur. J. 2019, 25, 2482–2485. doi:10.1002/chem.201806030
Return to citation in text: [1] -
Maroto, E. E.; Izquierdo, M.; Murata, M.; Filippone, S.; Komatsu, K.; Murata, Y.; Martín, N. Chem. Commun. 2014, 50, 740–742. doi:10.1039/c3cc46999a
Return to citation in text: [1] -
Maroto, E. E.; Mateos, J.; Garcia-Borràs, M.; Osuna, S.; Filippone, S.; Herranz, M. Á.; Murata, Y.; Solà, M.; Martín, N. J. Am. Chem. Soc. 2015, 137, 1190–1197. doi:10.1021/ja5108854
Return to citation in text: [1] -
Vidal, S.; Izquierdo, M.; Alom, S.; Garcia-Borràs, M.; Filippone, S.; Osuna, S.; Solà, M.; Whitby, R. J.; Martín, N. Chem. Commun. 2017, 53, 10993–10996. doi:10.1039/c7cc05987f
Return to citation in text: [1] -
Hashikawa, Y.; Murata, M.; Wakamiya, A.; Murata, Y. J. Am. Chem. Soc. 2016, 138, 4096–4104. doi:10.1021/jacs.5b12795
Return to citation in text: [1] -
Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Artigas, A.; Lledó, A.; Pla‐Quintana, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2017, 23, 15067–15072. doi:10.1002/chem.201702494
Return to citation in text: [1] -
Castro, E.; Artigas, A.; Pla-Quintana, A.; Roglans, A.; Liu, F.; Perez, F.; Lledó, A.; Zhu, X.-Y.; Echegoyen, L. Materials 2019, 12, 1314. doi:10.3390/ma12081314
Return to citation in text: [1] -
Taylor, R.; Wasserman, E.; Haddon, R. C.; Kroto, H. W. The pattern of additions to fullerenes. The Fullerenes; Cambridge University Press: Cambridge, UK, 1993; pp 87–102. doi:10.1017/cbo9780511622946.009
Return to citation in text: [1] -
Hirsch, A. The Chemistry of the Fullerenes; Thieme: Stuttgart, Germany, 1994.
Return to citation in text: [1] -
Fernández, I.; Solà, M.; Bickelhaupt, F. M. Chem. – Eur. J. 2013, 19, 7416–7422. doi:10.1002/chem.201300648
Return to citation in text: [1] -
Birkett, P. R.; Avent, A. G.; Darwish, A. D.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. J. Chem. Soc., Chem. Commun. 1995, 1869–1870. doi:10.1039/c39950001869
Return to citation in text: [1] -
Hashikawa, Y.; Sadai, S.; Murata, Y. Chem. Commun. 2023, 59, 7387–7390. doi:10.1039/d3cc01717f
Return to citation in text: [1] [2] -
Lou, N.; Li, Y.; Gan, L. Angew. Chem., Int. Ed. 2017, 56, 2403–2407. doi:10.1002/anie.201612054
Return to citation in text: [1] -
Sadai, S.; Hashikawa, Y.; Murata, Y. Org. Lett. 2023, 25, 2815–2819. doi:10.1021/acs.orglett.3c00726
Return to citation in text: [1] -
Murata, Y.; Maeda, S.; Murata, M.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 6702–6703. doi:10.1021/ja801753m
Return to citation in text: [1] [2] [3] -
Hashikawa, Y.; Shimizu, Y.; Murata, Y. Org. Lett. 2020, 22, 8624–8628. doi:10.1021/acs.orglett.0c03216
Return to citation in text: [1] -
Cerón, M. R.; Izquierdo, M.; Aghabali, A.; Valdez, J. A.; Ghiassi, K. B.; Olmstead, M. M.; Balch, A. L.; Wudl, F.; Echegoyen, L. J. Am. Chem. Soc. 2015, 137, 7502–7508. doi:10.1021/jacs.5b03768
Return to citation in text: [1] -
McKenzie, D. R.; Davis, C. A.; Cockayne, D. J. H.; Muller, D. A.; Vassallo, A. M. Nature 1992, 355, 622–624. doi:10.1038/355622a0
Return to citation in text: [1] -
Mestres, J.; Duran, M.; Solà, M. J. Phys. Chem. 1996, 100, 7449–7454. doi:10.1021/jp960312h
Return to citation in text: [1] -
Zhang, R.; Futagoishi, T.; Murata, M.; Wakamiya, A.; Murata, Y. J. Am. Chem. Soc. 2014, 136, 8193–8196. doi:10.1021/ja504054s
Return to citation in text: [1] -
Castro, E.; Fernandez-Delgado, O.; Artigas, A.; Zavala, G.; Liu, F.; Moreno-Vicente, A.; Rodríguez-Fortea, A.; Velasquez, J. D.; Poblet, J. M.; Echegoyen, L. J. Mater. Chem. C 2020, 8, 6813–6819. doi:10.1039/d0tc01382j
Return to citation in text: [1] -
Only the most stable of the two possible regioisomers of β-2a based on DFT calculations is depicted in Scheme 2 (see Figure S25 in Supporting Information File 1).
Return to citation in text: [1] -
Kozuch, S.; Shaik, S. J. Phys. Chem. A 2008, 112, 6032–6041. doi:10.1021/jp8004772
Return to citation in text: [1] -
Kozuch, S.; Shaik, S. Acc. Chem. Res. 2011, 44, 101–110. doi:10.1021/ar1000956
Return to citation in text: [1] -
Murata, M.; Maeda, S.; Morinaka, Y.; Murata, Y.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 15800–15801. doi:10.1021/ja8076846
Return to citation in text: [1] -
Morinaka, Y.; Tanabe, F.; Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2010, 46, 4532–4534. doi:10.1039/c0cc00113a
Return to citation in text: [1]
| 1. | Kroto, H. W.; Heath, J. R.; O’Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162–163. doi:10.1038/318162a0 |
| 14. | Jariwala, D.; Sangwan, V. K.; Lauhon, L. J.; Marks, T. J.; Hersam, M. C. Chem. Soc. Rev. 2013, 42, 2824–2860. doi:10.1039/c2cs35335k |
| 15. | Babu, S. S.; Möhwald, H.; Nakanishi, T. Chem. Soc. Rev. 2010, 39, 4021–4035. doi:10.1039/c000680g |
| 16. | Scarselli, M.; Castrucci, P.; De Crescenzi, M. J. Phys.: Condens. Matter 2012, 24, 313202. doi:10.1088/0953-8984/24/31/313202 |
| 39. | Birkett, P. R.; Avent, A. G.; Darwish, A. D.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. J. Chem. Soc., Chem. Commun. 1995, 1869–1870. doi:10.1039/c39950001869 |
| 40. | Hashikawa, Y.; Sadai, S.; Murata, Y. Chem. Commun. 2023, 59, 7387–7390. doi:10.1039/d3cc01717f |
| 41. | Lou, N.; Li, Y.; Gan, L. Angew. Chem., Int. Ed. 2017, 56, 2403–2407. doi:10.1002/anie.201612054 |
| 42. | Sadai, S.; Hashikawa, Y.; Murata, Y. Org. Lett. 2023, 25, 2815–2819. doi:10.1021/acs.orglett.3c00726 |
| 43. | Murata, Y.; Maeda, S.; Murata, M.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 6702–6703. doi:10.1021/ja801753m |
| 44. | Hashikawa, Y.; Shimizu, Y.; Murata, Y. Org. Lett. 2020, 22, 8624–8628. doi:10.1021/acs.orglett.0c03216 |
| 45. | Cerón, M. R.; Izquierdo, M.; Aghabali, A.; Valdez, J. A.; Ghiassi, K. B.; Olmstead, M. M.; Balch, A. L.; Wudl, F.; Echegoyen, L. J. Am. Chem. Soc. 2015, 137, 7502–7508. doi:10.1021/jacs.5b03768 |
| 9. | Kim, Y.; Cook, S.; Tuladhar, S. M.; Choulis, S. A.; Nelson, J.; Durrant, J. R.; Bradley, D. D. C.; Giles, M.; McCulloch, I.; Ha, C.-S.; Ree, M. Nat. Mater. 2006, 5, 197–203. doi:10.1038/nmat1574 |
| 10. | Collavini, S.; Delgado, J. L. Sustainable Energy Fuels 2018, 2, 2480–2493. doi:10.1039/c8se00254a |
| 11. | Jia, L.; Chen, M.; Yang, S. Mater. Chem. Front. 2020, 4, 2256–2282. doi:10.1039/d0qm00295j |
| 12. | Gopal, J.; Muthu, M.; Sivanesan, I. Polymers (Basel, Switz.) 2023, 15, 701. doi:10.3390/polym15030701 |
| 13. | Kausar, A. Polym.-Plast. Technol. Mater. 2023, 62, 618–631. doi:10.1080/25740881.2022.2121223 |
| 46. | McKenzie, D. R.; Davis, C. A.; Cockayne, D. J. H.; Muller, D. A.; Vassallo, A. M. Nature 1992, 355, 622–624. doi:10.1038/355622a0 |
| 7. | Montellano López, A.; Mateo-Alonso, A.; Prato, M. J. Mater. Chem. 2011, 21, 1305–1318. doi:10.1039/c0jm02386h |
| 8. | Canevet, D.; Pérez, E. M.; Martín, N. Angew. Chem., Int. Ed. 2011, 50, 9248–9259. doi:10.1002/anie.201101297 |
| 36. | Taylor, R.; Wasserman, E.; Haddon, R. C.; Kroto, H. W. The pattern of additions to fullerenes. The Fullerenes; Cambridge University Press: Cambridge, UK, 1993; pp 87–102. doi:10.1017/cbo9780511622946.009 |
| 37. | Hirsch, A. The Chemistry of the Fullerenes; Thieme: Stuttgart, Germany, 1994. |
| 38. | Fernández, I.; Solà, M.; Bickelhaupt, F. M. Chem. – Eur. J. 2013, 19, 7416–7422. doi:10.1002/chem.201300648 |
| 2. | Bakry, R.; Vallant, R. M.; Najam-ul-Haq, M.; Rainer, M.; Szabo, Z.; Huck, C. W.; Bonn, G. K. Int. J. Nanomed. 2007, 2, 639–649. |
| 3. | Castro, E.; Hernandez Garcia, A.; Zavala, G.; Echegoyen, L. J. Mater. Chem. B 2017, 5, 6523–6535. doi:10.1039/c7tb00855d |
| 4. | Panwar, N.; Soehartono, A. M.; Chan, K. K.; Zeng, S.; Xu, G.; Qu, J.; Coquet, P.; Yong, K.-T.; Chen, X. Chem. Rev. 2019, 119, 9559–9656. doi:10.1021/acs.chemrev.9b00099 |
| 5. | Ramos-Soriano, J.; Reina, J. J.; Illescas, B. M.; de la Cruz, N.; Rodríguez-Pérez, L.; Lasala, F.; Rojo, J.; Delgado, R.; Martín, N. J. Am. Chem. Soc. 2019, 141, 15403–15412. doi:10.1021/jacs.9b08003 |
| 6. | Cataldo, F.; Da Ros, T., Eds. Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes; Carbon Materials: Chemistry and Physics; Springer Netherlands: Dordrecht, Netherlands, 2008. doi:10.1007/978-1-4020-6845-4 |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 22. | Komatsu, K.; Murata, M.; Murata, Y. Science 2005, 307, 238–240. doi:10.1126/science.1106185 |
| 23. | Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2008, 6083–6094. doi:10.1039/b811738a |
| 24. | Hashikawa, Y.; Yasui, H.; Kurotobi, K.; Murata, Y. Mater. Chem. Front. 2018, 2, 206–213. doi:10.1039/c7qm00449d |
| 25. | Hashikawa, Y.; Murata, Y. Bull. Chem. Soc. Jpn. 2023, 96, 943–967. doi:10.1246/bcsj.20230135 |
| 28. | Hashikawa, Y.; Murata, Y. Chem. – Eur. J. 2019, 25, 2482–2485. doi:10.1002/chem.201806030 |
| 29. | Maroto, E. E.; Izquierdo, M.; Murata, M.; Filippone, S.; Komatsu, K.; Murata, Y.; Martín, N. Chem. Commun. 2014, 50, 740–742. doi:10.1039/c3cc46999a |
| 30. | Maroto, E. E.; Mateos, J.; Garcia-Borràs, M.; Osuna, S.; Filippone, S.; Herranz, M. Á.; Murata, Y.; Solà, M.; Martín, N. J. Am. Chem. Soc. 2015, 137, 1190–1197. doi:10.1021/ja5108854 |
| 31. | Vidal, S.; Izquierdo, M.; Alom, S.; Garcia-Borràs, M.; Filippone, S.; Osuna, S.; Solà, M.; Whitby, R. J.; Martín, N. Chem. Commun. 2017, 53, 10993–10996. doi:10.1039/c7cc05987f |
| 32. | Hashikawa, Y.; Murata, M.; Wakamiya, A.; Murata, Y. J. Am. Chem. Soc. 2016, 138, 4096–4104. doi:10.1021/jacs.5b12795 |
| 21. | Hummelen, J. C.; Prato, M.; Wudl, F. J. Am. Chem. Soc. 1995, 117, 7003–7004. doi:10.1021/ja00131a024 |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 34. | Artigas, A.; Lledó, A.; Pla‐Quintana, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2017, 23, 15067–15072. doi:10.1002/chem.201702494 |
| 35. | Castro, E.; Artigas, A.; Pla-Quintana, A.; Roglans, A.; Liu, F.; Perez, F.; Lledó, A.; Zhu, X.-Y.; Echegoyen, L. Materials 2019, 12, 1314. doi:10.3390/ma12081314 |
| 19. | Hirsch, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 1138–1141. doi:10.1002/anie.199311381 |
| 20. | Taylor, R. C. R. Chim. 2006, 9, 982–1000. doi:10.1016/j.crci.2006.01.004 |
| 17. | Ai, M.; Chen, M.; Yang, S. Chin. J. Chem. 2023, 41, 2337–2353. doi:10.1002/cjoc.202300105 |
| 18. | Paukov, M.; Kramberger, C.; Begichev, I.; Kharlamova, M.; Burdanova, M. Materials 2023, 16, 1276. doi:10.3390/ma16031276 |
| 26. | Stanisky, C. M.; Cross, R. J.; Saunders, M. J. Am. Chem. Soc. 2009, 131, 3392–3395. doi:10.1021/ja809831a |
| 27. |
Gao, R.; Liu, Z.; Liu, Z.; Liang, T.; Su, J.; Gan, L. Angew. Chem., Int. Ed. 2023, 62, e202300151. doi:10.1002/anie.202300151
and references cited therein. |
| 43. | Murata, Y.; Maeda, S.; Murata, M.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 6702–6703. doi:10.1021/ja801753m |
| 48. | Zhang, R.; Futagoishi, T.; Murata, M.; Wakamiya, A.; Murata, Y. J. Am. Chem. Soc. 2014, 136, 8193–8196. doi:10.1021/ja504054s |
| 47. | Mestres, J.; Duran, M.; Solà, M. J. Phys. Chem. 1996, 100, 7449–7454. doi:10.1021/jp960312h |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 40. | Hashikawa, Y.; Sadai, S.; Murata, Y. Chem. Commun. 2023, 59, 7387–7390. doi:10.1039/d3cc01717f |
| 43. | Murata, Y.; Maeda, S.; Murata, M.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 6702–6703. doi:10.1021/ja801753m |
| 53. | Murata, M.; Maeda, S.; Morinaka, Y.; Murata, Y.; Komatsu, K. J. Am. Chem. Soc. 2008, 130, 15800–15801. doi:10.1021/ja8076846 |
| 54. | Morinaka, Y.; Tanabe, F.; Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2010, 46, 4532–4534. doi:10.1039/c0cc00113a |
| 51. | Kozuch, S.; Shaik, S. J. Phys. Chem. A 2008, 112, 6032–6041. doi:10.1021/jp8004772 |
| 52. | Kozuch, S.; Shaik, S. Acc. Chem. Res. 2011, 44, 101–110. doi:10.1021/ar1000956 |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 33. | Artigas, A.; Pla‐Quintana, A.; Lledó, A.; Roglans, A.; Solà, M. Chem. – Eur. J. 2018, 24, 10653–10661. doi:10.1002/chem.201802298 |
| 49. | Castro, E.; Fernandez-Delgado, O.; Artigas, A.; Zavala, G.; Liu, F.; Moreno-Vicente, A.; Rodríguez-Fortea, A.; Velasquez, J. D.; Poblet, J. M.; Echegoyen, L. J. Mater. Chem. C 2020, 8, 6813–6819. doi:10.1039/d0tc01382j |
| 50. | Only the most stable of the two possible regioisomers of β-2a based on DFT calculations is depicted in Scheme 2 (see Figure S25 in Supporting Information File 1). |
© 2024 Castanyer et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.