Abstract
Meroterpenoids are hybrid compounds that are partially derived from terpenoids. This group of natural products displays large structural diversity, and many members exhibit beneficial biological activities. This mini-review highlights recent advances in the engineered biosynthesis of meroterpenoid compounds with C15 and C20 terpenoid moieties, with the reconstruction of fungal meroterpenoid biosynthetic pathways in heterologous expression hosts and the mutagenesis of key enzymes, including terpene cyclases and α-ketoglutarate (αKG)-dependent dioxygenases, that contribute to the structural diversity. Notable progress in genome sequencing has led to the discovery of many novel genes encoding these enzymes, while continued efforts in X-ray crystallographic analyses of these enzymes and the invention of AlphaFold2 have facilitated access to their structures. Structure-based mutagenesis combined with applications of unnatural substrates has further diversified the catalytic repertoire of these enzymes. The information in this review provides useful knowledge for the design of biosynthetic machineries to produce a variety of bioactive meroterpenoids.

Graphical Abstract
Introduction
Meroterpenoids are complex natural products with intricate skeletal structures, and are partially derived from terpenoids [1]. Many fungal meroterpenoids are composed of polyketide and terpenoid moieties. Examples of fungal meroterpenoids include mycophenolic acid (Figure 1, 1), which shows immunosuppressive activity and cell differentiation-inducing activity by inhibiting the IMP dehydrogenase involved in inosinic acid metabolism [2]; ascofuranone (Figure 1, 2), which exhibits antiparasitic activity by selectively inhibiting the oxidase of Plasmodium trypanosoma [3]; and pyripyropene (Figure 1, 3), which displays hyperlipidemia treatment activity through the inhibition of acyl-CoA: cholesterol acyltransferase [4]. In this mini-review, we focus on the fungal meroterpenoids biosynthesis, especially terpenonid cyclizations and post-cyclization modifications, which mostly contribute to the skeletal diversity. Several terpenoid cyclases and αKG-dependent dioxygenases will be discussed as examples of engineering biosynthetic pathways and key enzymes involved in fungal meroterpenoid biosynthesis. Furthermore, a construction of the artificial biosynthetic pathway composed of the fungal meroterpenoids pathway and the pathway from other species, in fungal host Aspergillus oryzae, will be also introduced.
![[1860-5397-20-50-1]](/bjoc/content/figures/1860-5397-20-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of bioactive fungal meroterpenoids.
Figure 1: Examples of bioactive fungal meroterpenoids.
Review
Standard reactions of fungal meroterpenoid biosynthesis
The skeletal diversity within this group of compounds arises from polyketide synthesis, prenyl transfer, terpenoid cyclizations, and post-cyclization modifications [5]. In these reactions, terpenoid cyclizations and post-cyclization modifications are especially important in fungal meroterpenoid biosynthesis, contributing to the structural diversity of this class of compounds. Fungal meroterpenoid cyclases (CYCs) are seven-membrane-spanning transporter-like enzymes that regulate the conformations of cationic intermediates, leading to the terpenoid structure of each product [6,7]. Particular attention has been paid to the biosynthesis of the compounds derived from farnesyl-DMOA (5) composed of 3,5-dimethylorsellinic acid (DMOA, 4) and the C15 terpenoid moiety due to their structural diversity (Figure 2).
![[1860-5397-20-50-2]](/bjoc/content/figures/1860-5397-20-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The diversity of DMOA-derived meroterpenoid biosyntheses.
Figure 2: The diversity of DMOA-derived meroterpenoid biosyntheses.
In 2012, the biosynthesis of terretonin, a meroterpenoid derived from compound 5 and produced by the filamentous fungus Aspergillus terreus, was investigated [8,9] using the heterologous expression host Aspergillus oryzae strain NSAR1, a filamentous fungus developed by multiple mutations of metabolic genes for ease of gene transfer and high substance production capabilities [10,11].
The expression of trt4 (polyketide synthase, PKS), trt2 (prenyltransferase, PT), trt5 (methyltransferase, MT), trt8 (flavin-dependent monooxygenase, FMO), and trt1 (meroterpenoid cyclase, CYC) in A. oryzae NSAR1 led to the production of preterretonin A (7) (Figure 2) [9]. These data indicated that Trt1 protonates the epoxide of (10'R)-epoxyfarnesyl-DMOA-3,5-methyl ester (6), leading to the cyclization of the terpenoid moiety in the chair–chair conformation, and catalyzes the deprotonation of H-9' of the resulting cation intermediate at C-4' to induce an acyl shift, forming the steroid-like structure of 7 with a 6-6-6-5 ring (Figure 2).
Swapping terpenoid cyclases in heterologous expression systems
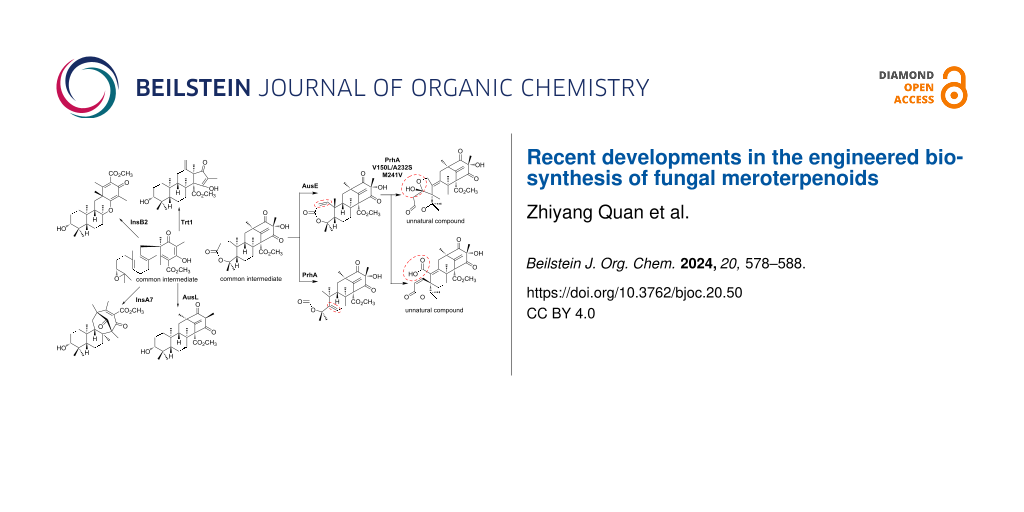
A search of the genome database for Trt1-homolog CYCs revealed the enzyme AusL (41% identity with Trt1) in Aspergillus nidulans, within the same phylogenetic clade. Expression of this enzyme in place of Trt1 resulted in the formation of the 6-6-6-6-membered ring protoaustinoid A (8) (Figure 2) [9]. In addition, the expression of the cyclase AdrI (38% identity with Trt1) from Penicillium chrysogenum produced the 6-6-6-5-membered andrastin E (9) (Figure 2) [8]. Like Trt1, AusL and AdrI create the common cation intermediate from 6, but they deprotonate the cationic intermediate from H-1' and H-11, respectively [12]. The differences in the structural bases of Trt1, AusL, and AdrI are quite intriguing, in that they have similar substrate binding modes but different regional specificities.
The rapid increase in genomic information has further expanded the number of available enzyme genes, thus increasing the potential for diversifying DMOA-derived meroterpenoid biosynthesis. Recently, a similar genome mining approach led to the discovery of new CYCs, InsA7 (32% identity with Trt1) and InsB2 (40% identity with Trt1) from Aspergillus insuetus [13]. These enzymes catalyze the formation of the terpenoid skeleton from 6, adopting chair–boat and chair–chair conformations to create two distinct 6-6-6-6-membered ring meroterpenoids: insuetusin A1 (12) and insuetusin B1 (10), respectively (Figure 2) [9]. Like other Trt1-type enzymes, InsB2 catalyzes the protonation of the epoxide, the formation of two six-membered rings in a chair–chair conformation, but the reaction finishes with the deprotonation of the hydroxy group at C-3 to produce compound 10. In contrast, InsA7 commonly initiates and terminates the reaction with the protonation of the epoxide and the deprotonation of OH-3, respectively, but it produces product 12 via a boat–chair conformation. Since all of the other Trt1-like cyclases catalyze the reaction with chair–chair stereocontrol, the structure basis of InsA7 should be unique. A structural comparison of InsB2 and InsA7 will help to clarify the differences in their structural bases.
Recent advances in artifical intelligence (AI) have even made the structural modeling of membrane-bound proteins possible. In a recent study, a structural model of AdrI was constructed by AlphaFold2 [14], and its product 9 was docked into the model [15]. In the complex structure, H76 and K187 are located close to the D ring of 9, and are expected to determine the selectivity of the product. The AdrI H76F/K187A variant was created, and produced the new compound 11, as expected. In this mutant, deprotonation occurs from the OH-3 of the folded intermediate, and the altered cyclization mode produces 11 (Figure 2).
Reconstitution based on the several meroterpenoid pathways in heterologous expression systems
Recently, the combinatorial biosynthesis of diterpene pyrones, derived from a pyrone skeleton and a C20 terpenoid skeleton, has been achieved by combining multiple biosynthetic pathways in a fungal heterologous expression host [16]. First, subA (PKS), subC (PT), subD (GGPP synthase), subE (FMO), and subB (CYC) were expressed in A. oryzae to produce the intermediate 15. Next, genes encoding a short-chain oxidoreductase (SDR), methyltransferase (MT), cytochrome P450 oxygenase (P450), and FMO from various fungi were additionally expressed in A. oryzae expessing subABCDE to produce 22 bioactive meroterpenoids, including the known antitumor compound subglutinol A (16) (Figure 3A and B) [17]. Among the novel compounds isolated from the production system, some exhibited intruiging pharmacological activities, such as antitumor (16), anti-HIV activity (17), and anti-Alzheimer's disease properties (18). Furthermore, FMO EsdpE, which epoxidizes the C-14, C-15 double bond of 13, was employed to produce 19, and the CYC EsdpB cyclizes in a chair–chair–chair conformation to form the 6-6-6 ring structure of 20 [18,19]. This difference in the epoxidation position represents a new point of biosynthetic diversity that will receive keen attention in the future.
![[1860-5397-20-50-3]](/bjoc/content/figures/1860-5397-20-50-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The combinatorial biosynthesis of diterpene pyrone meroterpenoids. The production of subglutinol A by SubABCDE + DpasF (A), the production of novel diterpene pyrones by SubABCDE + various tailoring enzymes (B), and the production of schearone A by SubACD + EsdpE + EsdpB (C).
Figure 3: The combinatorial biosynthesis of diterpene pyrone meroterpenoids. The production of subglutinol A ...
Production of pharmacologically active compounds by genetic engineering of terpenoid cyclization pathways
A single filamentous fungus reportedly synthesizes two meroterpenoid compounds via two different cyclization schemes. In the filamentous fungus Acremonium egypticum, both asochlorin (22) and ascofuranone (23) are synthesized from a common intermediate, ilicicolin A epoxide (21), but the backbones of the terpenoid moiety are different (Figure 4) [20]. In ascochlorin biosynthesis, intermediate 21 is accepted by AscF to form the hexanone ring of 22. The cyclization process is initiated by the protonation of the epoxide, leading to the formation of a cation at C-7. A cascade of migrations then occurs to form the final backbone: H-6 migrates to the cation C-7, the methyl group at C-13 shifts to C-6, H-10 migrates to C-11, and finally, deprotonation of the hydroxy group terminates the cyclization [20,21]. In ascofuranone biosynthesis, compound 21 is initially hydroxylated at C-8, and then the hydroxylated product is cyclized via epoxidation by AscI to form the tetrahydrofuran ring of 23. The biosynthetic pathways of 22 and 23 were elucidated through heterologous expression, gene disruption, and in vitro reactions. The ascF gene was disrupted with the aim toward the mass-production of 23, and a system for producing quantities greater than 500 mg/L was successfully established, thus achieving an industrial-level substance production system [20].
![[1860-5397-20-50-4]](/bjoc/content/figures/1860-5397-20-50-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The biosynthetic reaction from the common intermediate 21 to ascochlorin (22) and ascofuranone (23), respectively.
Figure 4: The biosynthetic reaction from the common intermediate 21 to ascochlorin (22) and ascofuranone (23)...
αKG-dependent dioxygenases – important enzymes involved in post-cyclization modifications
After terpenoid cyclization, skeletal modifications are performed by P450 monooxygenases, α-ketoglutarate (αKG)-dependent dioxygenases, isomerases, and acyltransferases [5,22-25]. In particular, the functions of the αKG-dependent dioxygenase have been analyzed in detail due to its relatively small molecular weight and the low costs of its cofactors: αKG, ascorbic acid, and iron ions. The αKG reacts with iron and molecular oxygen to form the highly reactive Fe(IV)=O via oxidative decarboxylation. This active molecular species withdraws a hydrogen atom, and the generated radical induces various reactions such as hydroxylation, unsaturation, epoxidation, halogenation, endoperoxidation, and C–C bond reconstruction, leading to the formation of diverse chemical structures [22,26-31].
Structure-based mutagenesis of αKG-dependent dioxygenases
The first αKG-dependent dioxygenases characterized in fungal meroterpenoid biosynthesis, AusE and PrhA, share high identity (76% identity to each other) and accept the meroterpenoid preaustinoid A1 (24) to yield the products preaustinoid A3 (27) and berkeleydione (28), respectively (Figure 5) [32,33]. In the initial reaction, AusE abstracts H-2 of 24, while PrhA abstracts H-5 of 24. PrhA and AusE form a double bond to yield preaustinoid A2 (25) and berkeleyone B (26), respectively (Figure 5). AusE abstracts H-5 of 25, forming a radical that initiates a sequential reaction to produce the spirolactone 27. In contrast, PrhA dehydrogenates H-1 of 26 to form the heptadiene 28. Comparing the active centers, only three amino acid residues differ between AusE and PrhA: L150(AusE)/V150(PrhA), S232(AusE)/A232(PrhA), and V241(AusE)/M241(PrhA). By swapping these residues, the catalytic activities of these enzymes were exchanged [34]. From 24, PrhA V150L/A232S produced 27, and AusE L150V/S232A produced 28. Surprisingly, PrhA V150L/A232S/M241V catalyzed additional oxidations of 27 to produce compounds 29 and 30, as unnatural products. As demonstrated in these reactions, the terpenoid skeleton undergoes significant structural changes due to radical formation through oxidase-induced hydrogen atom abstraction. Close examinations of the substrate complex structures of these αKG-dependent dioxygenases involved in these meroterpenoid oxidations revealed that the terpenoid moiety is held in a substrate pocket consisting of hydrophobic amino acids, and the polyketide moiety is retained by a loop structure serving as a lid. The conformation of the terpenoid moiety can be easily altered by mutations to the enzyme’s substrate retention site, thereby changing the position of hydrogen atom abstraction and the subsequent skeletal reorganization reaction mode, resulting in the synthesis of a variety of skeletons. Thus, the meroterpenoid αKG-dependent dioxygenase is a highly potent biosynthetic enzyme, and the structure of the product can be easily altered by mutagenesis.
![[1860-5397-20-50-5]](/bjoc/content/figures/1860-5397-20-50-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The multistep oxidations catalyzed by AusE and PrhA from the common intermediate 24.
Figure 5: The multistep oxidations catalyzed by AusE and PrhA from the common intermediate 24.
Application of diverse unnatural substrates to αKG-dependent dioxygenases
SptF, an α-KG dioxygenase, represents another example of structure-based engineering to create multiple products from a single enzyme reaction [35]. The natural substrates of SptF, andiconin D (31) and andilesin C (32) derived from andiconin, can be converted into emervaridone B (33) and anditomin (34), respectively (Figure 6). Subsequently, 33 can be further modified into emervaridone C (35), which can be oxidized to 37 or 38 (Figure 6). In contrast, 34 is converted to compounds 36a and 36b (Figure 6). These multistep reactions with several substrates illustrate the broad substrate specificity of SptF.
![[1860-5397-20-50-6]](/bjoc/content/figures/1860-5397-20-50-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Reactions of SptF with native substrates 31 and 32.
Figure 6: Reactions of SptF with native substrates 31 and 32.
To apply the high potential of SptF to generate a variety of oxidation products, meroterpenoids (39, 41, 42, 45, and 46) and terpenoids (49 and 52) were also used as unnatural substrates to construct a series of new compounds (40, 43, 44, 47, 48, 50, 51, and 53–55) (Figure 7A). In addition, a structure-based mutagenesis study of SptF was performed to further amplify its catalytic potential. Firstly, the hydrophobic residues Ile63, Phe133, and Ile231, which compose the substrate binding site of SptF, were mutated. As a result, SptF F133A oxidized 31 into 56, while I231A oxidized the same substrate 31 into compound 57 (Figure 7B). Then, the hydrophilic residues Asn65, Ser114, Thr148, and Asn150, which also line the cavity, were also mutated. Compound 31 was converted into 38 by SptF N150A, while the WT enzyme converted 31 into 37 and 38 (Figure 7B). These results exemplify the production of oxidized terpenoids and meroterpenoids through the use of non-natural substrates and the application of structure-based mutagenesis.
![[1860-5397-20-50-7]](/bjoc/content/figures/1860-5397-20-50-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: A) Reactions of SptF with unnatural substrates. B) Reactions of SptF variants with 31.
Figure 7: A) Reactions of SptF with unnatural substrates. B) Reactions of SptF variants with 31.
Evolution of αKG-dependent dioxygenase with saturated mutagenesis
As meroterpenoids readily change their reaction products depending on the conformation of the terpenoid skeleton, the regiospecificity of the oxidation reaction can be modified by introducing random mutations in the substrate-binding site of αKG-dependent dioxygenase. The αKG-dependent dioxygenase AndA withdraws H-12 of preandiloid C (58), and generates a radical leading to the construction of the bicyclo[2.2.2]octane skeleton of andiconin (59) (Figure 8) [36,37]. Two amino acid pairs, M119/N121 and A228/A230, in its substrate-binding pocket were mutated by saturation mutagenesis using degenerate primers, resulting in an enzyme that oxidizes C-5, thereby reacting with the electron between C-1 and C-2 [38]. In the future, such mutagenesis strategies will be applied to a wider range of oxidases to alter their reactivities and create an expanded range of products.
![[1860-5397-20-50-8]](/bjoc/content/figures/1860-5397-20-50-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The reaction of the αKG enzyme AndA and its variants generated via saturated mutagenesis.
Figure 8: The reaction of the αKG enzyme AndA and its variants generated via saturated mutagenesis.
Construction of artificial fungi–plant hybrid biosynthetic pathways
Furthermore, there are examples of the enzymatic synthesis of plant-derived pharmaceutical meroterpenoids through the heterologous expression of a combination of fungal and plant biosynthetic enzymes. The biosynthetic enzymes StbA (polyketide synthase, PKS) and StbC (prenyltransferase, PT) from the filamentous fungus Stachbotrys bisbyi were produced in A. oryzae to synthesize grifolic acid (60) (Figure 9) [39]. In addition, the gene encoding DCAS, a prenyl group oxidase from the plant Rhododendron dauricum, was additionally expressed in A. oryzae expressing stbAC after codon-optimization to produce daurichromenic acid (61) (Figure 9), a plant-derived meroterpenoid that exhibits anti-HIV activity [40]. Furthermore, by expressing AscD, a fungal halogenase from the ascochlorin biosynthetic pathway, the authors succeeded in biosynthesizing 62, an unnatural meroterpneoid, chloro-daurichromenic acid, halogenated at C-3 (Figure 9). Today, with the improvement of gene synthesis and expression technologies, genes from a wide variety of species are available, further increasing the potential for applications of biogenetic resources. In the future, advances in synthetic biology will enable the biosynthesis of bioactive compounds with still greater skeletal diversity.
![[1860-5397-20-50-9]](/bjoc/content/figures/1860-5397-20-50-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: The synthetic biological production of daurichromenic acid and its halogenated derivative.
Figure 9: The synthetic biological production of daurichromenic acid and its halogenated derivative.
Conclusion
In summary, many novel terpene cyclases and αKG-dependent dioxygenases were discovered by recent developments in genome mining approaches. Unique meroterpenoids have been generated by integrating these enzymes into meroterpenoid biosynthetic pathways, enzymatic engineering, and feeding with unnatural substrates. Enzyme crystal structure analysis and AI structure prediction also facilitated these investigations. Besides terpene cyclases and αKG-dependent dioxygenases, P450 monooxygenases and UbiA-type prenyltransferases have also expanded the variety of meroterpenoids [41,42]. These untapped membrane enzymes will be applicable for engineered biosynthesis, with structural models providing useful information for mutagenesis, as in the engineering of meroterpenoid cyclases [11] and fungal P450 oxygenases catalyzing cross-coupling between two aromatic rings [43]. In the future, further developments in bioinformatics, structural biology, and AI techniques will enable the design of biosynthetic enzymes and pathways to produce desired bioactive compounds, although understanding the chemistry catalyzed by individual enzymes will remain important. Furthermore, highly productive substance production systems will be developed through host, metabolome, and enzyme improvements by genome editing technology.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant Number JP21H02636 and JP22H05123), AMED (Grant Number JP21ak0101164) from Japan Science and Technology Agency, The Naito foundation, Nagase Science Technology Foundation, Yamada Science foundation, and The Mitsubishi Foundation.
Data Availability Statement
Data sharing is not applicable as no new data was generated or analyzed in this study.
References
-
Geris, R.; Simpson, T. J. Nat. Prod. Rep. 2009, 26, 1063–1094. doi:10.1039/b820413f
Return to citation in text: [1] -
Wu, J. C. Perspect. Drug Discovery Des. 1994, 2, 185–204. doi:10.1007/bf02171743
Return to citation in text: [1] -
Nakamura, K.; Fujioka, S.; Fukumoto, S.; Inoue, N.; Sakamoto, K.; Hirata, H.; Kido, Y.; Yabu, Y.; Suzuki, T.; Watanabe, Y.-i.; Saimoto, H.; Akiyama, H.; Kita, K. Parasitol. Int. 2010, 59, 560–564. doi:10.1016/j.parint.2010.07.006
Return to citation in text: [1] -
Tomoda, H.; Kim, Y. K.; Nishida, H.; Masuma, R.; Ōmura, S. J. Antibiot. 1994, 47, 148–153. doi:10.7164/antibiotics.47.148
Return to citation in text: [1] -
Matsuda, Y.; Abe, I. Nat. Prod. Rep. 2016, 33, 26–53. doi:10.1039/c5np00090d
Return to citation in text: [1] [2] -
Itoh, T.; Tokunaga, K.; Matsuda, Y.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. Nat. Chem. 2010, 2, 858–864. doi:10.1038/nchem.764
Return to citation in text: [1] -
Barra, L.; Abe, I. Nat. Prod. Rep. 2021, 38, 566–585. doi:10.1039/d0np00056f
Return to citation in text: [1] -
Itoh, T.; Tokunaga, K.; Radhakrishnan, E. K.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. ChemBioChem 2012, 13, 1132–1135. doi:10.1002/cbic.201200124
Return to citation in text: [1] [2] -
Matsuda, Y.; Awakawa, T.; Itoh, T.; Wakimoto, T.; Kushiro, T.; Fujii, I.; Ebizuka, Y.; Abe, I. ChemBioChem 2012, 13, 1738–1741. doi:10.1002/cbic.201200369
Return to citation in text: [1] [2] [3] [4] -
Jin, F.; Maruyama, J.; Juvvadi, P.; Arioka, M.; Kitamoto, K. Biosci., Biotechnol., Biochem. 2004, 68, 656–662. doi:10.1271/bbb.68.656
Return to citation in text: [1] -
Awakawa, T.; Abe, I. J. Fungi 2021, 7, 486–495. doi:10.3390/jof7060486
Return to citation in text: [1] [2] -
Matsuda, Y.; Awakawa, T.; Abe, I. Tetrahedron 2013, 69, 8199–8204. doi:10.1016/j.tet.2013.07.029
Return to citation in text: [1] -
Tang, J.; Matsuda, Y. Chem. Sci. 2022, 13, 10361–10369. doi:10.1039/d2sc02994d
Return to citation in text: [1] -
Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Nature 2021, 596, 583–589. doi:10.1038/s41586-021-03819-2
Return to citation in text: [1] -
Tang, J.; Matsuda, Y. Angew. Chem., Int. Ed. 2023, e202306046. doi:10.1002/anie.202306046
Return to citation in text: [1] -
Tsukada, K.; Shinki, S.; Kaneko, A.; Murakami, K.; Irie, K.; Murai, M.; Miyoshi, H.; Dan, S.; Kawaji, K.; Hayashi, H.; Kodama, E. N.; Hori, A.; Salim, E.; Kuraishi, T.; Hirata, N.; Kanda, Y.; Asai, T. Nat. Commun. 2020, 11, 1830. doi:10.1038/s41467-020-15664-4
Return to citation in text: [1] -
Lee, J. C.; Lobkovsky, E.; Pliam, N. B.; Strobel, G.; Clardy, J. J. Org. Chem. 1995, 60, 7076–7077. doi:10.1021/jo00127a001
Return to citation in text: [1] -
Morishita, Y.; Tsukada, K.; Murakami, K.; Irie, K.; Asai, T. J. Nat. Prod. 2022, 85, 384–390. doi:10.1021/acs.jnatprod.1c00973
Return to citation in text: [1] -
Xiao, Z.-H.; Dong, J.-Y.; Li, A.; Dai, J.-M.; Li, Y.-P.; Hu, Q.-F.; Shao, L.-D.; Matsuda, Y.; Wang, W.-G. Org. Chem. Front. 2022, 9, 1837–1843. doi:10.1039/d2qo00055e
Return to citation in text: [1] -
Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D. K.; Nagamune, K.; Ito, K.; Abe, I.; Kita, K. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8269–8274. doi:10.1073/pnas.1819254116
Return to citation in text: [1] [2] [3] -
Quan, Z.; Awakawa, T.; Wang, D.; Hu, Y.; Abe, I. Org. Lett. 2019, 21, 2330–2334. doi:10.1021/acs.orglett.9b00616
Return to citation in text: [1] -
Awakawa, T.; Mori, T.; Ushimaru, R.; Abe, I. Nat. Prod. Rep. 2023, 40, 46–61. doi:10.1039/d2np00014h
Return to citation in text: [1] [2] -
Zhang, X.; Guo, J.; Cheng, F.; Li, S. Nat. Prod. Rep. 2021, 38, 1072–1099. doi:10.1039/d1np00004g
Return to citation in text: [1] -
Mori, T.; Iwabuchi, T.; Hoshino, S.; Wang, H.; Matsuda, Y.; Abe, I. Nat. Chem. Biol. 2017, 13, 1066–1073. doi:10.1038/nchembio.2443
Return to citation in text: [1] -
Matsuda, Y.; Awakawa, T.; Mori, T.; Abe, I. Curr. Opin. Chem. Biol. 2016, 31, 1–7. doi:10.1016/j.cbpa.2015.11.001
Return to citation in text: [1] -
Gao, S.-S.; Naowarojna, N.; Cheng, R.; Liu, X.; Liu, P. Nat. Prod. Rep. 2018, 35, 792–837. doi:10.1039/c7np00067g
Return to citation in text: [1] -
Nakamura, H.; Matsuda, Y.; Abe, I. Nat. Prod. Rep. 2018, 35, 633–645. doi:10.1039/c7np00055c
Return to citation in text: [1] -
Ushimaru, R.; Abe, I. ACS Catal. 2023, 13, 1045–1076. doi:10.1021/acscatal.2c05247
Return to citation in text: [1] -
Matsuda, Y.; Bai, T.; Phippen, C. B. W.; Nødvig, C. S.; Kjærbølling, I.; Vesth, T. C.; Andersen, M. R.; Mortensen, U. H.; Gotfredsen, C. H.; Abe, I.; Larsen, T. O. Nat. Commun. 2018, 9, 2587. doi:10.1038/s41467-018-04983-2
Return to citation in text: [1] -
Mori, T.; Zhai, R.; Ushimaru, R.; Matsuda, Y.; Abe, I. Nat. Commun. 2021, 12, 4417. doi:10.1038/s41467-021-24685-6
Return to citation in text: [1] -
Li, X.; Awakawa, T.; Mori, T.; Ling, M.; Hu, D.; Wu, B.; Abe, I. J. Am. Chem. Soc. 2021, 143, 21425–21432. doi:10.1021/jacs.1c11548
Return to citation in text: [1] -
Matsuda, Y.; Awakawa, T.; Wakimoto, T.; Abe, I. J. Am. Chem. Soc. 2013, 135, 10962–10965. doi:10.1021/ja405518u
Return to citation in text: [1] -
Matsuda, Y.; Iwabuchi, T.; Fujimoto, T.; Awakawa, T.; Nakashima, Y.; Mori, T.; Zhang, H.; Hayashi, F.; Abe, I. J. Am. Chem. Soc. 2016, 138, 12671–12677. doi:10.1021/jacs.6b08424
Return to citation in text: [1] -
Nakashima, Y.; Mori, T.; Nakamura, H.; Awakawa, T.; Hoshino, S.; Senda, M.; Senda, T.; Abe, I. Nat. Commun. 2018, 9, 104. doi:10.1038/s41467-017-02371-w
Return to citation in text: [1] -
Tao, H.; Mori, T.; Chen, H.; Lyu, S.; Nonoyama, A.; Lee, S.; Abe, I. Nat. Commun. 2022, 13, No. 95. doi:10.1038/s41467-021-27636-3
Return to citation in text: [1] -
Matsuda, Y.; Wakimoto, T.; Mori, T.; Awakawa, T.; Abe, I. J. Am. Chem. Soc. 2014, 136, 15326–15336. doi:10.1021/ja508127q
Return to citation in text: [1] -
Nakashima, Y.; Mitsuhashi, T.; Matsuda, Y.; Senda, M.; Sato, H.; Yamazaki, M.; Uchiyama, M.; Senda, T.; Abe, I. J. Am. Chem. Soc. 2018, 140, 9743–9750. doi:10.1021/jacs.8b06084
Return to citation in text: [1] -
Mori, T.; Nakashima, Y.; Chen, H.; Hoshino, S.; Mitsuhashi, T.; Abe, I. Chem. Commun. 2022, 58, 5510–5513. doi:10.1039/d2cc00736c
Return to citation in text: [1] -
Li, C.; Matsuda, Y.; Gao, H.; Hu, D.; Yao, X. S.; Abe, I. ChemBioChem 2016, 17, 904–907. doi:10.1002/cbic.201600087
Return to citation in text: [1] -
Okada, M.; Saito, K.; Wong, C. P.; Li, C.; Wang, D.; Iijima, M.; Taura, F.; Kurosaki, F.; Awakawa, T.; Abe, I. Org. Lett. 2017, 19, 3183–3186. doi:10.1021/acs.orglett.7b01288
Return to citation in text: [1] -
Qi, B.-w.; Li, N.; Zhang, B.-b.; Zhang, Z.-k.; Wang, W.-j.; Liu, X.; Wang, J.; Awakawa, T.; Tu, P.-f.; Abe, I.; Shi, S.-p.; Li, J. Org. Lett. 2022, 24, 2526–2530. doi:10.1021/acs.orglett.2c00684
Return to citation in text: [1] -
Zhang, K.; Zhang, G.; Hou, X.; Ma, C.; Liu, J.; Che, Q.; Zhu, T.; Li, D. Org. Lett. 2022, 24, 2025–2029. doi:10.1021/acs.orglett.2c00495
Return to citation in text: [1] -
Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7
Return to citation in text: [1]
| 36. | Matsuda, Y.; Wakimoto, T.; Mori, T.; Awakawa, T.; Abe, I. J. Am. Chem. Soc. 2014, 136, 15326–15336. doi:10.1021/ja508127q |
| 37. | Nakashima, Y.; Mitsuhashi, T.; Matsuda, Y.; Senda, M.; Sato, H.; Yamazaki, M.; Uchiyama, M.; Senda, T.; Abe, I. J. Am. Chem. Soc. 2018, 140, 9743–9750. doi:10.1021/jacs.8b06084 |
| 38. | Mori, T.; Nakashima, Y.; Chen, H.; Hoshino, S.; Mitsuhashi, T.; Abe, I. Chem. Commun. 2022, 58, 5510–5513. doi:10.1039/d2cc00736c |
| 39. | Li, C.; Matsuda, Y.; Gao, H.; Hu, D.; Yao, X. S.; Abe, I. ChemBioChem 2016, 17, 904–907. doi:10.1002/cbic.201600087 |
| 1. | Geris, R.; Simpson, T. J. Nat. Prod. Rep. 2009, 26, 1063–1094. doi:10.1039/b820413f |
| 14. | Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Nature 2021, 596, 583–589. doi:10.1038/s41586-021-03819-2 |
| 4. | Tomoda, H.; Kim, Y. K.; Nishida, H.; Masuma, R.; Ōmura, S. J. Antibiot. 1994, 47, 148–153. doi:10.7164/antibiotics.47.148 |
| 15. | Tang, J.; Matsuda, Y. Angew. Chem., Int. Ed. 2023, e202306046. doi:10.1002/anie.202306046 |
| 3. | Nakamura, K.; Fujioka, S.; Fukumoto, S.; Inoue, N.; Sakamoto, K.; Hirata, H.; Kido, Y.; Yabu, Y.; Suzuki, T.; Watanabe, Y.-i.; Saimoto, H.; Akiyama, H.; Kita, K. Parasitol. Int. 2010, 59, 560–564. doi:10.1016/j.parint.2010.07.006 |
| 13. | Tang, J.; Matsuda, Y. Chem. Sci. 2022, 13, 10361–10369. doi:10.1039/d2sc02994d |
| 2. | Wu, J. C. Perspect. Drug Discovery Des. 1994, 2, 185–204. doi:10.1007/bf02171743 |
| 9. | Matsuda, Y.; Awakawa, T.; Itoh, T.; Wakimoto, T.; Kushiro, T.; Fujii, I.; Ebizuka, Y.; Abe, I. ChemBioChem 2012, 13, 1738–1741. doi:10.1002/cbic.201200369 |
| 9. | Matsuda, Y.; Awakawa, T.; Itoh, T.; Wakimoto, T.; Kushiro, T.; Fujii, I.; Ebizuka, Y.; Abe, I. ChemBioChem 2012, 13, 1738–1741. doi:10.1002/cbic.201200369 |
| 8. | Itoh, T.; Tokunaga, K.; Radhakrishnan, E. K.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. ChemBioChem 2012, 13, 1132–1135. doi:10.1002/cbic.201200124 |
| 10. | Jin, F.; Maruyama, J.; Juvvadi, P.; Arioka, M.; Kitamoto, K. Biosci., Biotechnol., Biochem. 2004, 68, 656–662. doi:10.1271/bbb.68.656 |
| 11. | Awakawa, T.; Abe, I. J. Fungi 2021, 7, 486–495. doi:10.3390/jof7060486 |
| 12. | Matsuda, Y.; Awakawa, T.; Abe, I. Tetrahedron 2013, 69, 8199–8204. doi:10.1016/j.tet.2013.07.029 |
| 43. | Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7 |
| 8. | Itoh, T.; Tokunaga, K.; Radhakrishnan, E. K.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. ChemBioChem 2012, 13, 1132–1135. doi:10.1002/cbic.201200124 |
| 9. | Matsuda, Y.; Awakawa, T.; Itoh, T.; Wakimoto, T.; Kushiro, T.; Fujii, I.; Ebizuka, Y.; Abe, I. ChemBioChem 2012, 13, 1738–1741. doi:10.1002/cbic.201200369 |
| 40. | Okada, M.; Saito, K.; Wong, C. P.; Li, C.; Wang, D.; Iijima, M.; Taura, F.; Kurosaki, F.; Awakawa, T.; Abe, I. Org. Lett. 2017, 19, 3183–3186. doi:10.1021/acs.orglett.7b01288 |
| 6. | Itoh, T.; Tokunaga, K.; Matsuda, Y.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. Nat. Chem. 2010, 2, 858–864. doi:10.1038/nchem.764 |
| 7. | Barra, L.; Abe, I. Nat. Prod. Rep. 2021, 38, 566–585. doi:10.1039/d0np00056f |
| 9. | Matsuda, Y.; Awakawa, T.; Itoh, T.; Wakimoto, T.; Kushiro, T.; Fujii, I.; Ebizuka, Y.; Abe, I. ChemBioChem 2012, 13, 1738–1741. doi:10.1002/cbic.201200369 |
| 41. | Qi, B.-w.; Li, N.; Zhang, B.-b.; Zhang, Z.-k.; Wang, W.-j.; Liu, X.; Wang, J.; Awakawa, T.; Tu, P.-f.; Abe, I.; Shi, S.-p.; Li, J. Org. Lett. 2022, 24, 2526–2530. doi:10.1021/acs.orglett.2c00684 |
| 42. | Zhang, K.; Zhang, G.; Hou, X.; Ma, C.; Liu, J.; Che, Q.; Zhu, T.; Li, D. Org. Lett. 2022, 24, 2025–2029. doi:10.1021/acs.orglett.2c00495 |
| 18. | Morishita, Y.; Tsukada, K.; Murakami, K.; Irie, K.; Asai, T. J. Nat. Prod. 2022, 85, 384–390. doi:10.1021/acs.jnatprod.1c00973 |
| 19. | Xiao, Z.-H.; Dong, J.-Y.; Li, A.; Dai, J.-M.; Li, Y.-P.; Hu, Q.-F.; Shao, L.-D.; Matsuda, Y.; Wang, W.-G. Org. Chem. Front. 2022, 9, 1837–1843. doi:10.1039/d2qo00055e |
| 16. | Tsukada, K.; Shinki, S.; Kaneko, A.; Murakami, K.; Irie, K.; Murai, M.; Miyoshi, H.; Dan, S.; Kawaji, K.; Hayashi, H.; Kodama, E. N.; Hori, A.; Salim, E.; Kuraishi, T.; Hirata, N.; Kanda, Y.; Asai, T. Nat. Commun. 2020, 11, 1830. doi:10.1038/s41467-020-15664-4 |
| 17. | Lee, J. C.; Lobkovsky, E.; Pliam, N. B.; Strobel, G.; Clardy, J. J. Org. Chem. 1995, 60, 7076–7077. doi:10.1021/jo00127a001 |
| 34. | Nakashima, Y.; Mori, T.; Nakamura, H.; Awakawa, T.; Hoshino, S.; Senda, M.; Senda, T.; Abe, I. Nat. Commun. 2018, 9, 104. doi:10.1038/s41467-017-02371-w |
| 35. | Tao, H.; Mori, T.; Chen, H.; Lyu, S.; Nonoyama, A.; Lee, S.; Abe, I. Nat. Commun. 2022, 13, No. 95. doi:10.1038/s41467-021-27636-3 |
| 22. | Awakawa, T.; Mori, T.; Ushimaru, R.; Abe, I. Nat. Prod. Rep. 2023, 40, 46–61. doi:10.1039/d2np00014h |
| 26. | Gao, S.-S.; Naowarojna, N.; Cheng, R.; Liu, X.; Liu, P. Nat. Prod. Rep. 2018, 35, 792–837. doi:10.1039/c7np00067g |
| 27. | Nakamura, H.; Matsuda, Y.; Abe, I. Nat. Prod. Rep. 2018, 35, 633–645. doi:10.1039/c7np00055c |
| 28. | Ushimaru, R.; Abe, I. ACS Catal. 2023, 13, 1045–1076. doi:10.1021/acscatal.2c05247 |
| 29. | Matsuda, Y.; Bai, T.; Phippen, C. B. W.; Nødvig, C. S.; Kjærbølling, I.; Vesth, T. C.; Andersen, M. R.; Mortensen, U. H.; Gotfredsen, C. H.; Abe, I.; Larsen, T. O. Nat. Commun. 2018, 9, 2587. doi:10.1038/s41467-018-04983-2 |
| 30. | Mori, T.; Zhai, R.; Ushimaru, R.; Matsuda, Y.; Abe, I. Nat. Commun. 2021, 12, 4417. doi:10.1038/s41467-021-24685-6 |
| 31. | Li, X.; Awakawa, T.; Mori, T.; Ling, M.; Hu, D.; Wu, B.; Abe, I. J. Am. Chem. Soc. 2021, 143, 21425–21432. doi:10.1021/jacs.1c11548 |
| 32. | Matsuda, Y.; Awakawa, T.; Wakimoto, T.; Abe, I. J. Am. Chem. Soc. 2013, 135, 10962–10965. doi:10.1021/ja405518u |
| 33. | Matsuda, Y.; Iwabuchi, T.; Fujimoto, T.; Awakawa, T.; Nakashima, Y.; Mori, T.; Zhang, H.; Hayashi, F.; Abe, I. J. Am. Chem. Soc. 2016, 138, 12671–12677. doi:10.1021/jacs.6b08424 |
| 20. | Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D. K.; Nagamune, K.; Ito, K.; Abe, I.; Kita, K. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8269–8274. doi:10.1073/pnas.1819254116 |
| 5. | Matsuda, Y.; Abe, I. Nat. Prod. Rep. 2016, 33, 26–53. doi:10.1039/c5np00090d |
| 22. | Awakawa, T.; Mori, T.; Ushimaru, R.; Abe, I. Nat. Prod. Rep. 2023, 40, 46–61. doi:10.1039/d2np00014h |
| 23. | Zhang, X.; Guo, J.; Cheng, F.; Li, S. Nat. Prod. Rep. 2021, 38, 1072–1099. doi:10.1039/d1np00004g |
| 24. | Mori, T.; Iwabuchi, T.; Hoshino, S.; Wang, H.; Matsuda, Y.; Abe, I. Nat. Chem. Biol. 2017, 13, 1066–1073. doi:10.1038/nchembio.2443 |
| 25. | Matsuda, Y.; Awakawa, T.; Mori, T.; Abe, I. Curr. Opin. Chem. Biol. 2016, 31, 1–7. doi:10.1016/j.cbpa.2015.11.001 |
| 20. | Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D. K.; Nagamune, K.; Ito, K.; Abe, I.; Kita, K. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8269–8274. doi:10.1073/pnas.1819254116 |
| 20. | Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D. K.; Nagamune, K.; Ito, K.; Abe, I.; Kita, K. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8269–8274. doi:10.1073/pnas.1819254116 |
| 21. | Quan, Z.; Awakawa, T.; Wang, D.; Hu, Y.; Abe, I. Org. Lett. 2019, 21, 2330–2334. doi:10.1021/acs.orglett.9b00616 |
© 2024 Quan and Awakawa; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.