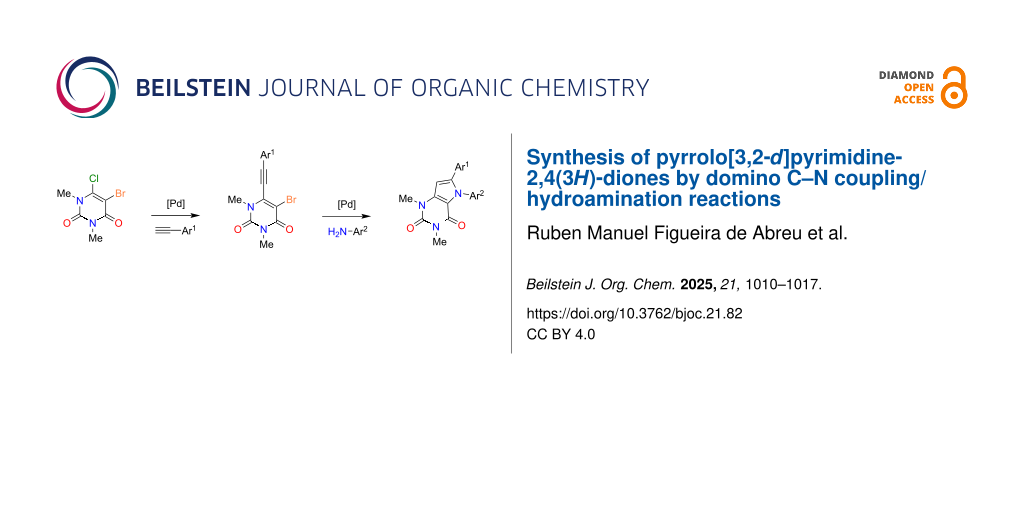
Abstract
A variety of pyrrolo[3,2-d]pyrimidine-2,4(3H)-diones were prepared by a combination of Sonogashira reaction and subsequent cyclization by domino C–N coupling/hydroamination reaction. The optical properties (UV–vis absorption and fluorescence) depend on the substitution pattern of the compounds.

Graphical Abstract
Introduction
Pyrimidines and purines are one of the most important heterocyclic compounds with prevalent biological functions. Both are found in nucleosides and their corresponding polymeric DNA and RNA, and hence are vital for life on Earth. The importance of these heteroaromatic derivatives has stimulated tremendous investigation towards the understanding of genetic information transmission as well as the synthesis of novel derivatives for medicinal applications [1-15]. For instance, deazapurines represent heterocyclic fused pyrimidine bases which have found special attention, due to their widespread occurrence in natural alkaloids exhibiting various biological properties. For example, cadeguomycin (A), tubercidin (B), and toyocamycin (C) show antibiotic properties, while batzelladine A (D), isolated from a Bahamian sponge, possesses anti-HIV and cancerostatic activities. Antiviral properties have been identified for sangivamycin (E), which was isolated from Streptomyces rimosus (Figure 1) [16-21].
![[1860-5397-21-82-1]](/bjoc/content/figures/1860-5397-21-82-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Development of drugs based on pyrrolopyrimidines: A: Cadeguomycin. B: Tubercidin. C: Toyocamycin. D: Batzelladine A. E: Sangivamycin. F: Pemetrexed. G: Immucillin H. H: TAK-285 (tyrosine kinase inhibitor).
Figure 1: Development of drugs based on pyrrolopyrimidines: A: Cadeguomycin. B: Tubercidin. C: Toyocamycin. D...
Consequently, several pyrrolopyrimidines have been synthesized to develop novel pharmaceuticals with improved biological properties. For instance, pemetrexed (F) is administered during palliative chemotherapy for advanced lung cancers [12]. Immucillin H (G) is currently in late clinical phases for the treatment of haematologic diseases, such as acute T-cell leukaemia, and TAK-285 (H) is a promising HER2/EGFR inhibitor which has been tested in a phase 1 trial on humans as an anticancer agent (Figure 1) [16]. Given the significance of deazapurines as biologically active lead compounds [22], we developed a new methodology for the synthesis of uracil-based pyrrolopyrimidine derivatives. The envisioned methodology combines a Sonogashira–Hagihara reaction and a one-pot process comprising a Buchwald–Hartwig coupling reaction followed by a hydroamination [23,24].
Results and Discussion
The bromination of 6-chloro-1,3-dimethyluracil (1) afforded, following a known procedure [25], 5-bromo-6-chloro-1,3-dimethyluracil (2) in 52% yield (Scheme 1). We previously reported Sonogashira reactions of the latter with various alkynes to give products 3a–d,g,h [26,27]. In this work, we extended the scope and prepared novel derivatives 3e and 3f. A nearly quantitative yield was obtained for product 3e derived from 3-tolylacetylene. However, the yield for compound 3f dropped to 60% because of the more sterically hindered 2-tolylacetylene.
![[1860-5397-21-82-i1]](/bjoc/content/inline/1860-5397-21-82-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3a–h. Conditions: i) Br2 (1.0 equiv), Ac2O (1.5 equiv), AcOH, 25 °C, 1 h [25]; ii) aryl acetylene (1.2 equiv), Pd(PPh3)Cl2 (5 mol %), CuI (5 mol %), NEt3 (10 equiv), DMSO, 25 °C, 6 h [26]. Yields of isolated products.
Scheme 1: Synthesis of 3a–h. Conditions: i) Br2 (1.0 equiv), Ac2O (1.5 equiv), AcOH, 25 °C, 1 h [25]; ii) aryl ac...
The domino C–N cross-coupling/hydroamination reaction of 3a–h with various anilines was studied next (Scheme 2) [28,29].
![[1860-5397-21-82-i2]](/bjoc/content/inline/1860-5397-21-82-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: C–N cross-coupling/hydroamination reaction.
Scheme 2: C–N cross-coupling/hydroamination reaction.
The conditions were optimized for the reaction of 3a with p-toluidine to give pyrrolo[3,2-d]pyrimidine-2,4(3H)-dione 4a (Table 1). For the first experiment, we chose Pd(OAc)2 (5 mol %) as the catalyst, XPhos (5 mol %) as the ligand and K3PO4 (3 equiv) as the base in DMA (100 °C, 15 hours), which previously proved to be efficient for related transformations [28,29]. However, only a yield of 15% of the desired product 4a was obtained after stirring for 15 hours, due to low conversion of the starting material. Subsequently, different mono- and bidentate ligands were tested. DPEphos was found to be the most potent ligand, leading to 43% isolated yield with full conversion of starting material and different byproducts derived from decomposition. Interestingly, no conversion of the starting material could be observed with the other ligands. In the following, different solvents, temperatures and bases were tested, but did not result in a further improvement of the yield.
Table 1: Optimization of the synthesis of 4a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-82-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
Ligand
(5 mol %) |
Base
(3 equiv) |
Solvent |
Temp
(°C) |
Yield
(%) |
| 1 | XPhos | K3PO4 | DMA | 100 | 15 |
| 2 | Xantphos | K3PO4 | DMA | 100 | – |
| 3 | DPEphos | K3PO4 | DMA | 100 | 43 |
| 4 | Dppf | K3PO4 | DMA | 100 | – |
| 5 | CataCXium A | K3PO4 | DMA | 100 | – |
| 6 | Ruphos | K3PO4 | DMA | 100 | – |
| 7 | P(t-Bu)3·HBF4 | K3PO4 | DMA | 100 | – |
| 8 | DPEphos | K3PO4 | toluene | 100 | 25 |
| 9 | DPEphos | K3PO4 | 1,4-dioxane | 100 | 34 |
| 10 | DPEphos | NaOt-Bu | DMA | 100 | 14 |
| 11 | DPEphos | KOt-Bu | DMA | 100 | 15 |
| 12 | DPEphos | Cs2CO3 | DMA | 100 | 9 |
| 13 | DPEphos | KN(SiMe3)2 | DMA | 100 | – |
| 14 | DPEphos | K3PO4 | DMA | 120 | 15 |
With the optimized conditions in hand, the scope of the reaction was studied. The cyclization of alkynylated uracils 3a–h with various anilines afforded pyrrolo[3,2-d]pyrimidine-2,4(3H)-diones 4a–m in moderate to good yields (Scheme 3). Various functional groups attached to the aniline, such as OMe, F, CF3, Me, and Br, were tolerated and did not greatly differ in terms of yield. With respect to substituents located at the alkynyl moiety, diminished yields were obtained for N,N-dimethylaminophenyl-substituted derivatives 4k and 4l and for m-tolyl-substituted compound 4h. No conversion was observed for starting materials 3g,h containing electron-withdrawing substituents located at the phenylacetylene moiety (products 4n,p). In addition, no conversion was observed for 3-methylaniline (product 4o).
![[1860-5397-21-82-i3]](/bjoc/content/inline/1860-5397-21-82-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4a–m. Conditions: Pd(OAc)2 (5 mol %), DPEphos (5 mol %), K3PO4 (3 equiv), DMA, 100 °C, 15 h. Yields of isolated products.
Scheme 3: Synthesis of 4a–m. Conditions: Pd(OAc)2 (5 mol %), DPEphos (5 mol %), K3PO4 (3 equiv), DMA, 100 °C,...
The photophysical properties of selected pyrrolo[3,2-d]pyrimidine-2,4(3H)-diones 4 were investigated by steady-state absorption and photoluminescence spectroscopy (Figure 2, Table 2). The wavelength and intensity of absorption and emission depended on the substitution pattern. The methyl-substituted compound 4a showed one broad absorption band at ≈290 nm. A similar absorption feature, but slightly bathochromically shifted, was observed for the thienyl-substituted derivative 4m. The strongly electron-donating N,N-dimethylaminophenyl group (products 4k,l) resulted in segmentation into two absorption bands accompanied by a strongly red-shifted lower energy absorption band. With respect to the phenyl group attached to the pyrrole nitrogen, the presence of the electron-withdrawing CF3 group led to a slight hypsochromic shift (4j). In the case of compound 4l, containing a 4-trifluoromethylphenyl group attached to the nitrogen, no significant shift of the absorption was observed as compared to compound 4k containing a 4-tolyl group. However, the presence of a CF3 group resulted in a reduction of the extinction coefficient of the absorption band.
![[1860-5397-21-82-2]](/bjoc/content/figures/1860-5397-21-82-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis absorption (left) and emission (right, λex = 300 nm) spectra of compounds 4a, 4j, 4k, 4l, and 4m in dichloromethane (c = 1·10−5 M).
Figure 2: UV–vis absorption (left) and emission (right, λex = 300 nm) spectra of compounds 4a, 4j, 4k, 4l, an...
Table 2: Photophysical data of 4a, 4j, 4k, 4l, and 4m in dichloromethane (c = 1·10−5 M) at 20 °C.
| 4a | 4j | 4k | 4l | 4m | |
|
λ1,abs (nm)
ελ1·104 (M−1 cm−1) |
293
1.5 |
278
1.7 |
289
1.4 |
297
1.4 |
299
1.7 |
|
λ2,abs (nm)
ελ2·104 (M−1 cm−1) |
335
2.6 |
339
1.7 |
|||
|
λ1,em400 (nm)
λ2,em400 (nm) |
377
462a |
380 | 418 | 436 | 364 |
| Φb | 9% | 0.1% | 83% | 71% | 4% |
aShoulder in the spectrum. bFor the excitation wavelength λex = 300 nm; fluorescence standard: quinine sulfate in H2SO4 (0.05 M) (Φ = 0.52) [30].
The emission spectrum of 4a consisted of a strong emission band at 377 nm with a weak shoulder expiring to higher wavelengths. However, this shoulder was less distinct for compounds 4m and 4j and not detectable for 4k and 4l, containing the N,N-dimethylaminophenyl group. In contrast to the absorption spectra, in the case of the emission spectra, the presence of a thienyl substituent (compound 4m) led to a hypsochromic shift, while the emission band of 4j was slightly red-shifted. The N,N-dimethylaminophenyl-substituted compounds 4k and 4l showed the strongest bathochromic shifts of the emission spectra, which might be due to the occurrence of donor–acceptor interactions between the electron-deficient uracil and the amino group. The corresponding fluorescence quantum yields were also strongly affected by the substitution pattern of the pyrrolouracils. Compounds 4k and 4l show very high fluorescence quantum yields of 83% and 71%, respectively, what might be reasoned by the strong donor ability of the NMe2-functional groups of those compounds. In contrast, compounds 4a and 4m showed only weak fluorescence with quantum yields of 9 and 4%, respectively. Compound 4j exhibited almost no emission (Φ = 0.1%). [30].
Conclusion
In summary, we developed a new methodology for the synthesis of pyrrolo[3,2-d]pyrimidine-2,4(3H)-diones based on a domino C–N coupling/hydroamination reaction of readily available alkynylated uracils with anilines. The optimized reaction conditions allowed for the employment of various functional groups. The products revealed fluorescence properties which were influenced by the substitution pattern. Electron-donating N,N-dimethylaminophenyl substituents led to bathochromically shifted absorption and emission spectra accompanied by strongly elevated fluorescence quantum yields (up to 83%). Further studies will be devoted to the synthesis of novel polycyclic uracil derivatives with potential biological activities.
Supporting Information
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 6.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Tanaka, K.; Sugawa, T.; Nakamori, R.; Sanno, Y.; Ando, Y.; Imai, K.-i. Chem. Pharm. Bull. 1964, 12, 1024–1030. doi:10.1248/cpb.12.1024
Return to citation in text: [1] -
Montgomery, J. A.; Hewson, K. J. Org. Chem. 1965, 30, 1528–1532. doi:10.1021/jo01016a046
Return to citation in text: [1] -
Javahershenas, R.; Khalafy, J. Heterocycl. Commun. 2018, 24, 37–41. doi:10.1515/hc-2017-0187
Return to citation in text: [1] -
Noell, C. W.; Robins, R. K. J. Heterocycl. Chem. 1964, 1, 34–41. doi:10.1002/jhet.5570010108
Return to citation in text: [1] -
Tkachenko, Yu. N.; Tsupak, E. B.; Pozharskii, A. F. Chem. Heterocycl. Compd. 2000, 36, 307–310. doi:10.1007/bf02256868
Return to citation in text: [1] -
Ogura, H.; Sakaguchi, M.; Takeda, K. Chem. Pharm. Bull. 1972, 20, 404–408. doi:10.1248/cpb.20.404
Return to citation in text: [1] -
Nasr, M. N.; Gineinah, M. M. Arch. Pharm. (Weinheim, Ger.) 2002, 335, 289. doi:10.1002/1521-4184(200208)335:6<289::aid-ardp289>3.0.co;2-z
Return to citation in text: [1] -
Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; Kamiguchi, H.; Tanaka, T.; Habuka, N.; Sogabe, S.; Yano, J.; Aertgeerts, K.; Kamiyama, K. J. Med. Chem. 2011, 54, 8030–8050. doi:10.1021/jm2008634
Return to citation in text: [1] -
Kawakita, Y.; Banno, H.; Ohashi, T.; Tamura, T.; Yusa, T.; Nakayama, A.; Miki, H.; Iwata, H.; Kamiguchi, H.; Tanaka, T.; Habuka, N.; Sogabe, S.; Ohta, Y.; Ishikawa, T. J. Med. Chem. 2012, 55, 3975–3991. doi:10.1021/jm300185p
Return to citation in text: [1] -
Temburnikar, K. W.; Ross, C. R.; Wilson, G. M.; Balzarini, J.; Cawrse, B. M.; Seley-Radtke, K. L. Bioorg. Med. Chem. 2015, 23, 4354–4363. doi:10.1016/j.bmc.2015.06.025
Return to citation in text: [1] -
Bottegoni, G.; Veronesi, M.; Bisignano, P.; Kacker, P.; Favia, A. D.; Cavalli, A. ChemMedChem 2016, 11, 1259–1263. doi:10.1002/cmdc.201500521
Return to citation in text: [1] -
Pathania, S.; Rawal, R. K. Eur. J. Med. Chem. 2018, 157, 503–526. doi:10.1016/j.ejmech.2018.08.023
Return to citation in text: [1] [2] -
Cawrse, B. M.; Robinson, N. M.; Lee, N. C.; Wilson, G. M.; Seley-Radtke, K. L. Molecules 2019, 24, 2656. doi:10.3390/molecules24142656
Return to citation in text: [1] -
Grahner, B.; Winiwarter, S.; Lanzner, W.; Müller, C. E. J. Med. Chem. 1994, 37, 1526–1534. doi:10.1021/jm00036a019
Return to citation in text: [1] -
Esteban-Gamboa, A.; Balzarini, J.; Esnouf, R.; De Clercq, E.; Camarasa, M.-J.; Pérez-Pérez, M.-J. J. Med. Chem. 2000, 43, 971–983. doi:10.1021/jm9911377
Return to citation in text: [1] -
De Coen, L. M.; Heugebaert, T. S. A.; García, D.; Stevens, C. V. Chem. Rev. 2016, 116, 80–139. doi:10.1021/acs.chemrev.5b00483
Return to citation in text: [1] [2] -
Yuan, B. D.; Wu, R. T.; Sato, I.; Okabe, T.; Suzuki, H.; Nishimura, T.; Tanaka, N. J. Antibiot. 1985, 38, 642–648. doi:10.7164/antibiotics.38.642
Return to citation in text: [1] -
Patil, A. D.; Kumar, N. V.; Kokke, W. C.; Bean, M. F.; Freyer, A. J.; de Brosse, C.; Mai, S.; Truneh, A.; Carte, B. J. Org. Chem. 1995, 60, 1182–1188. doi:10.1021/jo00110a021
Return to citation in text: [1] -
Acs, G.; Reich, E.; Mori, M. Proc. Natl. Acad. Sci. U. S. A. 1964, 52, 493–501. doi:10.1073/pnas.52.2.493
Return to citation in text: [1] -
Nishimura, H.; Katagiri, K.; Sato, K.; Mayama, M.; Shimaoka, N. J. Antibiot., Ser. A 1956, 9, 60–62. doi:10.11554/antibioticsa.9.2_60
Return to citation in text: [1] -
Saneyoshi, M.; Tokuzen, R.; Fukuoka, F. Gann 1965, 56, 219–222. doi:10.20772/cancersci1959.56.2_219
Return to citation in text: [1] -
Baraldi, P. G.; Romagnoli, R.; Saponaro, G.; Aghazadeh Tabrizi, M.; Baraldi, S.; Pedretti, P.; Fusi, C.; Nassini, R.; Materazzi, S.; Geppetti, P.; Preti, D. Bioorg. Med. Chem. 2012, 20, 1690–1698. doi:10.1016/j.bmc.2012.01.020
Return to citation in text: [1] -
Tang, Z.-Y.; Hu, Q.-S. Adv. Synth. Catal. 2006, 348, 846–850. doi:10.1002/adsc.200606022
Return to citation in text: [1] -
Lavery, C. B.; McDonald, R.; Stradiotto, M. Chem. Commun. 2012, 48, 7277–7279. doi:10.1039/c2cc33071g
Return to citation in text: [1] -
Pfleiderer, W.; Deiss, H. Isr. J. Chem. 1968, 6, 603–614. doi:10.1002/ijch.196800078
Return to citation in text: [1] [2] -
de Abreu, R. M. F.; Brockmann, T.; Villinger, A.; Ehlers, P.; Langer, P. Beilstein J. Org. Chem. 2024, 20, 898–911. doi:10.3762/bjoc.20.80
Return to citation in text: [1] [2] -
de Abreu, R. M. F.; Ehlers, P.; Langer, P. Beilstein J. Org. Chem. 2024, 20, 2708–2719. doi:10.3762/bjoc.20.228
Return to citation in text: [1] -
Do, H. H.; Hauptmann, R.; Villinger, A.; Surkus, A.-E.; Lochbrunner, S.; Ehlers, P.; Langer, P. Tetrahedron 2017, 73, 3407–3414. doi:10.1016/j.tet.2017.04.012
Return to citation in text: [1] [2] -
Langer, P. Synlett 2022, 33, 1596–1606. doi:10.1055/s-0041-1738384
Return to citation in text: [1] [2] -
Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. doi:10.1039/b912178a
Return to citation in text: [1] [2]
| 1. | Tanaka, K.; Sugawa, T.; Nakamori, R.; Sanno, Y.; Ando, Y.; Imai, K.-i. Chem. Pharm. Bull. 1964, 12, 1024–1030. doi:10.1248/cpb.12.1024 |
| 2. | Montgomery, J. A.; Hewson, K. J. Org. Chem. 1965, 30, 1528–1532. doi:10.1021/jo01016a046 |
| 3. | Javahershenas, R.; Khalafy, J. Heterocycl. Commun. 2018, 24, 37–41. doi:10.1515/hc-2017-0187 |
| 4. | Noell, C. W.; Robins, R. K. J. Heterocycl. Chem. 1964, 1, 34–41. doi:10.1002/jhet.5570010108 |
| 5. | Tkachenko, Yu. N.; Tsupak, E. B.; Pozharskii, A. F. Chem. Heterocycl. Compd. 2000, 36, 307–310. doi:10.1007/bf02256868 |
| 6. | Ogura, H.; Sakaguchi, M.; Takeda, K. Chem. Pharm. Bull. 1972, 20, 404–408. doi:10.1248/cpb.20.404 |
| 7. | Nasr, M. N.; Gineinah, M. M. Arch. Pharm. (Weinheim, Ger.) 2002, 335, 289. doi:10.1002/1521-4184(200208)335:6<289::aid-ardp289>3.0.co;2-z |
| 8. | Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; Kamiguchi, H.; Tanaka, T.; Habuka, N.; Sogabe, S.; Yano, J.; Aertgeerts, K.; Kamiyama, K. J. Med. Chem. 2011, 54, 8030–8050. doi:10.1021/jm2008634 |
| 9. | Kawakita, Y.; Banno, H.; Ohashi, T.; Tamura, T.; Yusa, T.; Nakayama, A.; Miki, H.; Iwata, H.; Kamiguchi, H.; Tanaka, T.; Habuka, N.; Sogabe, S.; Ohta, Y.; Ishikawa, T. J. Med. Chem. 2012, 55, 3975–3991. doi:10.1021/jm300185p |
| 10. | Temburnikar, K. W.; Ross, C. R.; Wilson, G. M.; Balzarini, J.; Cawrse, B. M.; Seley-Radtke, K. L. Bioorg. Med. Chem. 2015, 23, 4354–4363. doi:10.1016/j.bmc.2015.06.025 |
| 11. | Bottegoni, G.; Veronesi, M.; Bisignano, P.; Kacker, P.; Favia, A. D.; Cavalli, A. ChemMedChem 2016, 11, 1259–1263. doi:10.1002/cmdc.201500521 |
| 12. | Pathania, S.; Rawal, R. K. Eur. J. Med. Chem. 2018, 157, 503–526. doi:10.1016/j.ejmech.2018.08.023 |
| 13. | Cawrse, B. M.; Robinson, N. M.; Lee, N. C.; Wilson, G. M.; Seley-Radtke, K. L. Molecules 2019, 24, 2656. doi:10.3390/molecules24142656 |
| 14. | Grahner, B.; Winiwarter, S.; Lanzner, W.; Müller, C. E. J. Med. Chem. 1994, 37, 1526–1534. doi:10.1021/jm00036a019 |
| 15. | Esteban-Gamboa, A.; Balzarini, J.; Esnouf, R.; De Clercq, E.; Camarasa, M.-J.; Pérez-Pérez, M.-J. J. Med. Chem. 2000, 43, 971–983. doi:10.1021/jm9911377 |
| 22. | Baraldi, P. G.; Romagnoli, R.; Saponaro, G.; Aghazadeh Tabrizi, M.; Baraldi, S.; Pedretti, P.; Fusi, C.; Nassini, R.; Materazzi, S.; Geppetti, P.; Preti, D. Bioorg. Med. Chem. 2012, 20, 1690–1698. doi:10.1016/j.bmc.2012.01.020 |
| 16. | De Coen, L. M.; Heugebaert, T. S. A.; García, D.; Stevens, C. V. Chem. Rev. 2016, 116, 80–139. doi:10.1021/acs.chemrev.5b00483 |
| 12. | Pathania, S.; Rawal, R. K. Eur. J. Med. Chem. 2018, 157, 503–526. doi:10.1016/j.ejmech.2018.08.023 |
| 30. | Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. doi:10.1039/b912178a |
| 16. | De Coen, L. M.; Heugebaert, T. S. A.; García, D.; Stevens, C. V. Chem. Rev. 2016, 116, 80–139. doi:10.1021/acs.chemrev.5b00483 |
| 17. | Yuan, B. D.; Wu, R. T.; Sato, I.; Okabe, T.; Suzuki, H.; Nishimura, T.; Tanaka, N. J. Antibiot. 1985, 38, 642–648. doi:10.7164/antibiotics.38.642 |
| 18. | Patil, A. D.; Kumar, N. V.; Kokke, W. C.; Bean, M. F.; Freyer, A. J.; de Brosse, C.; Mai, S.; Truneh, A.; Carte, B. J. Org. Chem. 1995, 60, 1182–1188. doi:10.1021/jo00110a021 |
| 19. | Acs, G.; Reich, E.; Mori, M. Proc. Natl. Acad. Sci. U. S. A. 1964, 52, 493–501. doi:10.1073/pnas.52.2.493 |
| 20. | Nishimura, H.; Katagiri, K.; Sato, K.; Mayama, M.; Shimaoka, N. J. Antibiot., Ser. A 1956, 9, 60–62. doi:10.11554/antibioticsa.9.2_60 |
| 21. | Saneyoshi, M.; Tokuzen, R.; Fukuoka, F. Gann 1965, 56, 219–222. doi:10.20772/cancersci1959.56.2_219 |
| 30. | Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. doi:10.1039/b912178a |
| 25. | Pfleiderer, W.; Deiss, H. Isr. J. Chem. 1968, 6, 603–614. doi:10.1002/ijch.196800078 |
| 28. | Do, H. H.; Hauptmann, R.; Villinger, A.; Surkus, A.-E.; Lochbrunner, S.; Ehlers, P.; Langer, P. Tetrahedron 2017, 73, 3407–3414. doi:10.1016/j.tet.2017.04.012 |
| 29. | Langer, P. Synlett 2022, 33, 1596–1606. doi:10.1055/s-0041-1738384 |
| 26. | de Abreu, R. M. F.; Brockmann, T.; Villinger, A.; Ehlers, P.; Langer, P. Beilstein J. Org. Chem. 2024, 20, 898–911. doi:10.3762/bjoc.20.80 |
| 27. | de Abreu, R. M. F.; Ehlers, P.; Langer, P. Beilstein J. Org. Chem. 2024, 20, 2708–2719. doi:10.3762/bjoc.20.228 |
| 28. | Do, H. H.; Hauptmann, R.; Villinger, A.; Surkus, A.-E.; Lochbrunner, S.; Ehlers, P.; Langer, P. Tetrahedron 2017, 73, 3407–3414. doi:10.1016/j.tet.2017.04.012 |
| 29. | Langer, P. Synlett 2022, 33, 1596–1606. doi:10.1055/s-0041-1738384 |
| 25. | Pfleiderer, W.; Deiss, H. Isr. J. Chem. 1968, 6, 603–614. doi:10.1002/ijch.196800078 |
| 23. | Tang, Z.-Y.; Hu, Q.-S. Adv. Synth. Catal. 2006, 348, 846–850. doi:10.1002/adsc.200606022 |
| 24. | Lavery, C. B.; McDonald, R.; Stradiotto, M. Chem. Commun. 2012, 48, 7277–7279. doi:10.1039/c2cc33071g |
| 26. | de Abreu, R. M. F.; Brockmann, T.; Villinger, A.; Ehlers, P.; Langer, P. Beilstein J. Org. Chem. 2024, 20, 898–911. doi:10.3762/bjoc.20.80 |
© 2025 Figueira de Abreu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.