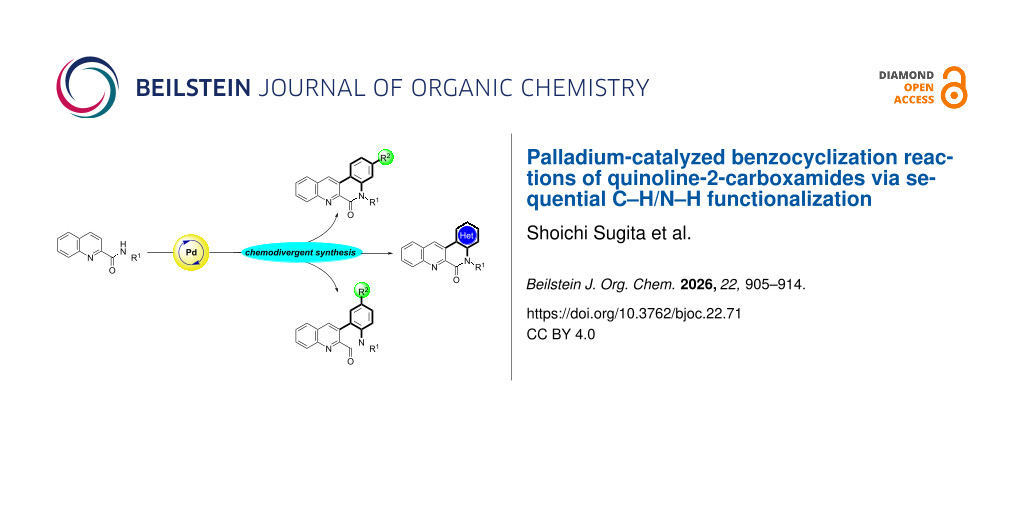
Abstract
A novel benzocyclization protocol has been developed for the synthesis of quinoline-fused lactams by palladium-catalyzed sequential C–H/N–H functionalization of quinoline-2-carboxamides and 1,2-dihaloarenes. The reaction proceeds at the C–H bond on the quinoline adjacent to the amide group and at the amide N–H bond in the presence of 10 mol % Pd(OAc)2 in o-xylene as a solvent to afford the cyclized product in 34% yield. The yield increases to 81% when the reaction is carried out with 80 mol % P(4-MeOC6H4)3 as a ligand and with an increased catalyst loading of 20 mol %. The reaction affords lactams in up to 83% yield using amides containing various functional groups and substituted 1-bromo-2-iodobenzenes. Furthermore, 1,2-dibromo heteroarenes, such as benzothiophene and pyridine, undergo annulation to give the corresponding heterocycle-fused compounds. The high chemoselectivity of the 1,2-dihaloarene functional groups is confirmed in this reaction, thus enabling divergent synthesis of various multifused heterocyclic systems.

Graphical Abstract
Introduction
Cyclic structures comprising nitrogen-containing multifused rings are extremely important because such heterocycle-fused cyclic structures [1,2] are found in various advanced materials [3,4] and biologically essential molecules [5-7]. Quinoline is a particularly intriguing moiety in biologically active compounds (e.g., natural products used for medicines, quinine, and quinidine [5-7]) and synthesized pharmaceutical agents (e.g., quinolone antibiotics [8]). Moreover, quinoline-2-carboxamide derivatives are used as ligands in organic synthesis owing to their high metal affinity [9,10]. It is therefore expected that annulation of the amide moiety in quinoline-2-carboxamides would extend their functionality as biologically active structures, ligands, and extractants.
Transition-metal-catalyzed coupling reactions are crucial for constructing carbon–carbon and carbon–heteroatom bonds. The classical coupling reactions, such as Kumada–Tamao–Corriu coupling [11-13], Sonogashira coupling [14,15], Negishi coupling [16,17], Migita–Kosugi–Stille coupling [18-20], Suzuki–Miyaura coupling [21,22], and Hiyama coupling [23,24] involve carbon–halogen and carbon–metal species (Scheme 1a). Fagnou and co-workers reported direct arylation reactions with palladium(II) acetate to synthesize biaryl compounds via concerted metalation–deprotonation (CMD; Scheme 1b) [25-27]. Buchwald, Hartwig, and co-workers explored carbon–nitrogen coupling reactions that allow facile preparation of aromatic amines (Scheme 1c) [28-31]. In general, intramolecular C–H arylation reactions in the presence of a transition-metal catalyst have been reported extensively in recent years. These reactions enable an efficient formation of fused-ring systems [32-35]. Additionally, intramolecular C–H arylation reactions with N-heteroaromatics can be used to synthesize various functional molecules that serve as ligands for metal extraction [36-41].
![[1860-5397-22-71-i1]](/bjoc/content/inline/1860-5397-22-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Various transition-metal-catalyzed coupling reactions involving aryl halides.
Scheme 1: Various transition-metal-catalyzed coupling reactions involving aryl halides.
Our group developed a cyclization reaction for the intramolecular C–H arylation with N-heterocycles, such as phenanthroline and quinoline, containing amide groups (Scheme 2a) [38,40]. These reactions efficiently provide the corresponding annulation products; however, it remains difficult to selectively obtain a variety of substituent positions following cyclization. Moreover, these methodologies necessitate the preparation of annulation precursors through o-brominated aniline derivatives [36-41]. Furthermore, the reported methodologies for synthesizing N-heterocycle-fused lactams are characterized by either low efficiency or protracted processes [42,43]. On the other hand, benzocyclization reactions of aryl carboxamides offer a method for synthesizing chemodivergent products from a single substrate. Importantly, these reactions are controlled by the different reactivities of the halogen atoms in the reagent structures. Several carbocycle C–H/N–H activated benzocyclizations have already been reported [44-54], although the reaction mechanism with π-deficient N-heteroaromatics has not been elucidated (Scheme 2b). It is therefore valuable to investigate the differences in reactivity between C–H and N–H for intermolecular arylation in the presence of transition-metal catalysts. This can reveal their selectivity in terms of reaction position(s) in chemodivergent synthesis (Scheme 2c). The present report explores benzocyclization reactions involving sequential C–H/N–H functionalization by a palladium catalyst.
![[1860-5397-22-71-i2]](/bjoc/content/inline/1860-5397-22-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic strategy for preparing fused lactams.
Scheme 2: Synthetic strategy for preparing fused lactams.
Results and Discussion
First, the C–H/N–H annulation reaction between quinoline-2-carboxamide 1a and 1-bromo-2-iodobenzene (2a) was tested. When 1a was treated with 1.0 equiv of 2a, 10 mol % Pd(OAc)2, 40 mol % PPh3, and 3.0 equiv of Cs2CO3 in dimethylformamide (DMF) at 140 °C, the desired lactam 3aa was obtained in 19% yield (Table 1, entry 1). This result indicated that the anticipated C–H and N–H intermolecular–intramolecular coupling reactions occurred. When PdCl2(PPh3)2 was used instead of Pd(OAc)2 as a catalyst in the absence of PPh3, 3aa was afforded in 10% yield (Table 1, entry 2). When Pd(OCOCF3)2 was used as the catalyst, the desired product was obtained in 39% yield (Table 1, entry 3). Using any base other than Cs2CO3 resulted in lower yields, thus confirming that Cs2CO3 was the optimal base for this reaction (Table 1, entries 4–6). Increasing the concentration of 1a from 0.1 to 1.0 M led to an increased yield, even if Pd(OAc)2 was used as the catalyst (Table 1, entry 7). When the reaction temperature was reduced to 110 °C, 120 °C, or 130 °C, product 3aa was obtained in lower yields in all cases compared with entry 7 (Table 1, entries 8–10). Using o-xylene as the solvent gave results similar to those obtained with DMF (Table 1, entry 11). Notably, the reaction using o-xylene as the solvent supported a good mass balance of 1a and 3aa. Therefore, the coupling reaction conditions were further optimized using o-xylene as the solvent. The ligand effect was examined, and the results indicated that electron-donating ligands were more efficient than electron-withdrawing ligands, particularly in the context of tris(4-methoxyphenyl)phosphine (P(4-MeOC6H4)3) (Table 1, entries 12–15). Additionally, the annulation product was detected in less than 1% yield in the presence of bulky ligands, such as tri(o-tolyl)phosphine (Table 1, entry 16). Next, the relative amounts of reagents were optimized for this annulation reaction (Table 1, entries 17–19). Ultimately, it was determined that the optimal reagent quantities were 20 mol % Pd(OAc)2, 80 mol % P(4-MeOC6H4)3, and 2.0 equiv of 2a, which afforded the product 3aa in 81% yield (Table 1, entry 19).
Table 1: Investigation of this C–H/N–H functionalization reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-22-71-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Catalyst (mol %) | Ligand (mol %) | Base (equiv) | 2a (equiv) | Solvent (M) | Yield (%)a | |
| 3aa | 1a | ||||||
| 1 | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (0.1) | 19 | 52 |
| 2 | PdCl2(PPh3)2 (10) | none | Cs2CO3 (3.0) | 1.0 | DMF (0.1) | 10 | 85 |
| 3 | Pd(OCOCF3)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (0.1) | 39 | 32 |
| 4 | Pd(OAc)2 (10) | PPh3 (40) | K2CO3 (3.0) | 1.0 | DMF (0.1) | 6 | 72 |
| 5 | Pd(OAc)2 (10) | PPh3 (40) | t-BuOK (3.0) | 1.0 | DMF (0.1) | 0 | quant. |
| 6 | Pd(OAc)2 (10) | PPh3 (40) | t-BuONa (3.0) | 1.0 | DMF (0.1) | 9 | 63 |
| 7 | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (1.0) | 46 | 28 |
| 8b | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (1.0) | 4 | 64 |
| 9c | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (1.0) | 19 | 61 |
| 10d | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | DMF (1.0) | 40 | 32 |
| 11 | Pd(OAc)2 (10) | PPh3 (40) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | 34 | 58 |
| 12 | Pd(OAc)2 (10) | PPh3 (100) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | 39 | 30 |
| 13 | Pd(OAc)2 (10) | P(p-tolyl)3 (40) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | 35 | 30 |
| 14 | Pd(OAc)2 (10) | P(4-MeOC6H4)3 (40) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | 44 | 33 |
| 15 | Pd(OAc)2 (10) | P(4-FC6H4)3 (40) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | 21 | 55 |
| 16 | Pd(OAc)2 (10) | P(o-tolyl)3 (40) | Cs2CO3 (3.0) | 1.0 | o-xylene (1.0) | <1 | quant. |
| 17e | Pd(OAc)2 (10) | P(4-MeOC6H4)3 (40) | Cs2CO3 (3.0) | 1.0 + 1.0 | o-xylene (1.0) | 53 | 15 |
| 18 | Pd(OAc)2 (10) | P(4-MeOC6H4)3 (80) | Cs2CO3 (3.0) | 2.0 | o-xylene (1.0) | 45 | 34 |
| 19 | Pd(OAc)2 (20) | P(4-MeOC6H4)3 (80) | Cs2CO3 (3.0) | 2.0 | o-xylene (1.0) | 81f | N.D.g |
aThe yields were determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard; bthe reaction was performed at 110 °C; cthe reaction was performed at 120 °C; dthe reaction was performed at 130 °C; eafter stirring for 20 hours at 140 °C, an additional equivalent of 2a was added to the reaction system, and the reaction was allowed to proceed for another 20 hours at 140 °C; fisolated yield; gnot detected.
The optimized conditions were used to investigate the substrate scope of this C–H/N–H functionalized reaction. First, the substituents at the amido group of quinoline-2-carboxamides 1 were examined (Table 2). The C–H/N–H functionalization reactions afforded good to excellent yields, regardless of the presence of a non-substituted phenyl group or various substituents at the 4-position of the phenyl moiety (Table 2, entries 1–5). In contrast, the quinoline-2-carboxamide containing a 4-nitrophenyl group gave the corresponding product in a low yield (Table 2, entry 6). This result indicated that an increased acidity of the N–H proton of the amides, caused by substituent groups, could potentially inhibit the annulation reaction. Similarly, amides bearing mesityl, 2-nitrophenyl, or 2-methoxyphenyl groups afforded the corresponding products in low yields due to steric hindrance (Table 2, entries 7–9). The reaction with benzyl-substituted carboxamide also resulted in a low yield owing to the lower acidity of the amide proton compared with aromatic amides (Table 2, entry 10). Based on these results quinoline carboxamides 1 bearing aromatic substituents, which avoid steric effects and induce moderate acidity of the amide N–H proton, exhibit higher propensity for reaction in comparison to aliphatic amides.
Table 2: Investigation of the substrate scope and substituent limitations of amide groups for the C–H/N–H functionalization reaction.
![[Graphic 2]](/bjoc/content/inline/1860-5397-22-71-i7.svg?max-width=637&scale=1.0)
|
||
| Entry | R | Yield (%) |
| 1 | Ph | 81 (3ba) |
| 2 | 4-MeOC6H4 | 68 (3ca) |
| 3 | 4-t-BuC6H4 | 72 (3da) |
| 4 | 4-CNC6H4 | 67 (3ea) |
| 5 | 4-ClC6H4 | 75 (3fa) |
| 6 | 4-NO2C6H4 | 18 (3ga) |
| 7 | mesityl | 21 (3ha) |
| 8 | 2-NO2C6H4 | 14 (3ia) |
| 9a | 2-MeOC6H4 | 17 (3ja) |
| 10a | Bn | 21 (3ka) |
aThe reaction time was prolonged to 96 h.
Next, the C–H/N–H functionalization reaction was evaluated using various 1,2-dihaloarenes 2 (Table 3). When 1-bromo-2-iodo-5-methylbenzene (2b) was reacted with 1a, the corresponding cycloadduct 3ab was obtained in good yield (Table 3, entry 1). In contrast, 1-bromo-2-iodo-4-methylbenzene (2c) afforded the annulation product 3ac in low yield (Table 3, entry 2). However, the desired products 3ab and 3ac were obtained in excellent yields when the reaction period was prolonged from 20 to 96 h. Although relatively low product yields were obtained when using dihaloarenes 2d and 2e, both containing a tert-butyl group, after 20 h, the corresponding products 3ad and 3ae were obtained in good to excellent yields when the reaction time was prolonged to 96 h (Table 3, entries 3 and 4). Similar trends were observed in the reactions involving 2-bromo-1-iodo-4-methoxybenzene (2f) and 1-bromo-2-iodo-4-methoxybenzene (1g) (Table 3, entries 5 and 6). The oxidative addition of the palladium catalyst to the C–I bond in these electron-donating group-containing bromo(iodo)benzenes occurred slowly, requiring an extended reaction time to reach completion. Particularly, when 2-bromo-1-iodobenzenes containing electron-donating groups at the C4 position were utilized in this reaction, the yields of the products 3 would be decreased due to the increased electron density of bromo(iodo)benzenes which results in an inhibition of the catalyst’s oxidative addition to the C–I bond. Notably, the substrates containing electron-withdrawing groups, such as cyano or nitro groups, resulted in low yields, even after longer reaction times (Table 3, entries 7 and 8). These results were attributed to homo-coupling of 1-bromo-2-iodobenzenes or protonation of activated haloarenes and deactivation of the palladium catalyst. It is therefore indicated that bromo(iodo)benzenes with high electron density utilized in this reaction require significantly longer reaction times to achieve good to excellent yields of products 3, whereas the use of bromo(iodo)benzenes with low electron density leads to their decomposition, which results in decreased yields of product 3. Finally, the coupling reactions with heteroarenes, such as 2,3-dibromobenzothiophene (2j) and 2,3-dibromopyridine (2k), afforded the corresponding products in moderate yields with high regioselectivity (Table 3, entries 9 and 10).
Table 3: Investigation of the dihaloarene substrate scope for the C–H/N–H functionalization reaction.
![[Graphic 3]](/bjoc/content/inline/1860-5397-22-71-i8.svg?max-width=637&scale=1.0)
|
||
| Entry | 2 | Product (yield, %) |
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-22-71-i9.svg?max-width=637&scale=1.0)
2b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-22-71-i10.svg?max-width=637&scale=1.0)
3ab (63, 79a) |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-22-71-i11.svg?max-width=637&scale=1.0)
2c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-22-71-i12.svg?max-width=637&scale=1.0)
3ac (17, 83a) |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-22-71-i13.svg?max-width=637&scale=1.0)
2d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-22-71-i14.svg?max-width=637&scale=1.0)
3ad (28, 55a) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-22-71-i15.svg?max-width=637&scale=1.0)
2e |
![[Graphic 11]](/bjoc/content/inline/1860-5397-22-71-i16.svg?max-width=637&scale=1.0)
3ae (18, 81a) |
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-22-71-i17.svg?max-width=637&scale=1.0)
2f |
![[Graphic 13]](/bjoc/content/inline/1860-5397-22-71-i18.svg?max-width=637&scale=1.0)
3af (32, 51a) |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-22-71-i19.svg?max-width=637&scale=1.0)
2g |
![[Graphic 15]](/bjoc/content/inline/1860-5397-22-71-i20.svg?max-width=637&scale=1.0)
3ag (24, 82a) |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-22-71-i21.svg?max-width=637&scale=1.0)
2h |
![[Graphic 17]](/bjoc/content/inline/1860-5397-22-71-i22.svg?max-width=637&scale=1.0)
3ah (26, 21a) |
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-22-71-i23.svg?max-width=637&scale=1.0)
2i |
![[Graphic 19]](/bjoc/content/inline/1860-5397-22-71-i24.svg?max-width=637&scale=1.0)
3ai (7, 7a) |
| 9 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-22-71-i25.svg?max-width=637&scale=1.0)
2j |
![[Graphic 21]](/bjoc/content/inline/1860-5397-22-71-i26.svg?max-width=637&scale=1.0)
3aj (21) |
| 10 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-22-71-i27.svg?max-width=637&scale=1.0)
2k |
![[Graphic 23]](/bjoc/content/inline/1860-5397-22-71-i28.svg?max-width=637&scale=1.0)
3ak (31) |
aThese yields were obtained when the reaction time was prolonged to 96 h.
The structures of the isomeric products were confirmed by nuclear Overhauser effect spectroscopy (NOESY, see Supporting Information File 1). First, the structures of lactams 3ab, 3ac, and 3ad were determined (Figure 1a–c, respectively). The products 3ab and 3ac, which were synthesized from 2b and 2c, were detected as the corresponding isomers, with the methyl group attached to the C3 or C2 position of products 3ab and 3ac, respectively. In the case of product 3ad, derived from 1-bromo-2-iodobenzene 2d, which contains a tert-butyl group in the C5 position, a similar substitution pattern was detected as for compound 3ad derived from 1-bromo-2-iodo-5-methylbenzene (2b). Additionally, during the coupling reaction carried out with 5-tert-butyl-1-bromo-2-iodobenzene, Int-1ad species was detected as a reaction intermediate, as confirmed by 1H NMR and mass spectra (Scheme 3; see also Supporting Information File 1). This result indicated that the C–C bond was formed first in the reaction. The structures of other products were inferred from their 1H NMR spectra, while the structures of products 3aj and 3ak were also determined by NOESY (Figure 1d and e).
![[1860-5397-22-71-1]](/bjoc/content/figures/1860-5397-22-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Nuclear Overhauser effect (NOE) correlations in products (a) 3ab, (b) 3ac, (c) 3ad, (d) 3aj, and (e) 3ak.
Figure 1: Nuclear Overhauser effect (NOE) correlations in products (a) 3ab, (b) 3ac, (c) 3ad, (d) 3aj, and (e...
![[1860-5397-22-71-i3]](/bjoc/content/inline/1860-5397-22-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Detection of the intermediate Int-1ad in the annulation reaction of 1a with 1-bromo-5-tert-butyl-2-iodobenzene (2d).
Scheme 3: Detection of the intermediate Int-1ad in the annulation reaction of 1a with 1-bromo-5-tert-butyl-2-...
These results suggested that the carbon–iodine bond in 1-bromo-2-iodobenzene was involved in the formation of the C–C bond, while the carbon–bromine bond was involved in the formation of the C–N bond (Scheme 4). Accordingly, a plausible reaction mechanism is proposed in Scheme 5. First, the activated palladium(0) catalyst is inserted into the carbon–iodine bond of the 1-bromo-2-iodoarene 2 via oxidative addition. The intermediate Int-2 then undergoes ligand exchange from iodine and phosphine to acetate and quinoline-2-carboxamide to generate intermediate Int-3. Then, through a concerted metalation-deprotonation (CMD) process, a carbon–palladium bond is formed to give the palladacycle intermediate Int-4. Next, reductive elimination between the quinoline and arene moieties forms the C–C bond to give intermediate Int-1 and regenerates the palladium(0) catalyst. Another oxidative addition to the carbon–bromine bond of Int-1 generates Int-5, and a nitrogen–palladium bond is formed to afford the seven membered palladacycle intermediate Int-6. Finally, this intermediate undergoes reductive elimination between the nitrogen and carbon atoms to provide the lactam product 3.
![[1860-5397-22-71-i4]](/bjoc/content/inline/1860-5397-22-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Stepwise formation of C–C and C–N bonds during the annulation reaction.
Scheme 4: Stepwise formation of C–C and C–N bonds during the annulation reaction.
![[1860-5397-22-71-i5]](/bjoc/content/inline/1860-5397-22-71-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible reaction mechanism for the sequential C–H/N–H functionalization reaction.
Scheme 5: Plausible reaction mechanism for the sequential C–H/N–H functionalization reaction.
Conclusion
This study explored a novel palladium-catalyzed C–H/N–H activated annulation reaction for the synthesis of quinoline-fused lactams. The annulation reaction afforded the desired products in up to 83% yield. The reaction displayed broad tolerance for various substituents. Moreover, the reaction demonstrated high chemoselectivity because it proceeds via initial C–C bond formation at C–I, followed by C–N bond formation at C–Br. Thus, the positions of substituents on the products are controlled based on the position of the substituents in the 1-bromo-2-iodobenzene substrates, thus providing facile and efficient access to chemodivergent products. The developed reaction protocol is expected to be applicable to the synthesis of functional materials and bioactive molecules.
Supporting Information
| Supporting Information File 1: Experimental details and copies of 1H and 13C{1H} NMR spectra. | ||
| Format: PDF | Size: 14.6 MB | Download |
Acknowledgements
We thank Suzanne Adam, Ph.D, from Edanz (https://jp.edanz.com/ac) for editing a draft of the manuscript.
Data Availability Statement
Additional research data generated and analyzed during this study is not shared.
References
-
Ganguly, A. K.; Wang, C. H.; David, M.; Bartner, P.; Chan, T. M. Tetrahedron Lett. 2002, 43, 6865–6868. doi:10.1016/s0040-4039(02)01537-x
Return to citation in text: [1] -
Das, S.; Kundu, S.; Metya, A.; Maji, M. S. Chem. Sci. 2024, 15, 13466–13474. doi:10.1039/d4sc03438d
Return to citation in text: [1] -
Guo, Z.; Yan, C.; Zhu, W.-H. Angew. Chem., Int. Ed. 2020, 59, 9812–9825. doi:10.1002/anie.201913249
Return to citation in text: [1] -
Yu, C. P.; Yamamoto, A.; Kumagai, S.; Takeya, J.; Okamoto, T. Angew. Chem., Int. Ed. 2023, 62, e202206417. doi:10.1002/anie.202206417
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2002, 19, 742–760. doi:10.1039/b104971m
Return to citation in text: [1] [2] -
Shang, X.-F.; Morris‐Natschke, S. L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Med. Res. Rev. 2018, 38, 775–828. doi:10.1002/med.21466
Return to citation in text: [1] [2] -
Yadav, T. T.; Murahari, M.; Peters, G. J.; YC, M. Eur. J. Med. Chem. 2022, 239, 114527. doi:10.1016/j.ejmech.2022.114527
Return to citation in text: [1] [2] -
Pham, T. D. M.; Ziora, Z. M.; Blaskovich, M. A. T. Med. Chem. Commun. 2019, 10, 1719–1739. doi:10.1039/c9md00120d
Return to citation in text: [1] -
Patra, S.; Hirose, T.; Himeda, Y. Inorg. Chim. Acta 2025, 585, 122769. doi:10.1016/j.ica.2025.122769
Return to citation in text: [1] -
Kumar, R.; Majumder, S.; Singh, C. P.; Samanta, C.; Newalkar, B. L.; Krishnamurty, S.; Das, R. K. ChemSusChem 2025, 18, e202502039. doi:10.1002/cssc.202502039
Return to citation in text: [1] -
Tamao, K.; Sumitani, K.; Kumada, M. J. Am. Chem. Soc. 1972, 94, 4374–4376. doi:10.1021/ja00767a075
Return to citation in text: [1] -
Corriu, R. J. P.; Masse, J. P. J. Chem. Soc., Chem. Commun. 1972, 144a. doi:10.1039/c3972000144a
Return to citation in text: [1] -
Heravi, M. M.; Zadsirjan, V.; Hajiabbasi, P.; Hamidi, H. Monatsh. Chem. 2019, 150, 535–591. doi:10.1007/s00706-019-2364-6
Return to citation in text: [1] -
Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084. doi:10.1039/c1cs15071e
Return to citation in text: [1] -
King, A. O.; Okukado, N.; Negishi, E.-i. J. Chem. Soc., Chem. Commun. 1977, 683–684. doi:10.1039/c39770000683
Return to citation in text: [1] -
Muzammil; Zahoor, A. F.; Parveen, B.; Javed, S.; Akhtar, R.; Tabassum, S. Chem. Pap. 2024, 78, 3399–3430. doi:10.1007/s11696-024-03369-7
Return to citation in text: [1] -
Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Migita, T. Chem. Lett. 1977, 6, 301–302. doi:10.1246/cl.1977.301
Return to citation in text: [1] -
Milstein, D.; Stille, J. K. J. Am. Chem. Soc. 1978, 100, 3636–3638. doi:10.1021/ja00479a077
Return to citation in text: [1] -
Srivastav, N.; Singh, R.; Kaur, V. RSC Adv. 2015, 5, 62202–62213. doi:10.1039/c5ra09960a
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866
Return to citation in text: [1] -
Lennox, A. J. J.; Lloyd-Jones, G. C. Chem. Soc. Rev. 2014, 43, 412–443. doi:10.1039/c3cs60197h
Return to citation in text: [1] -
Hatanaka, Y.; Hiyama, T. J. Org. Chem. 1988, 53, 918–920. doi:10.1021/jo00239a056
Return to citation in text: [1] -
Hiyama, T.; Minami, Y.; Mori, A. Transition-Metal-Catalyzed Cross-coupling of Organosilicon Compounds. In Organosilicon Chemistry; Hiyama, T.; Oestreich, M., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp 271–332. doi:10.1002/9783527814787.ch9
Return to citation in text: [1] -
Campeau, L.-C.; Parisien, M.; Leblanc, M.; Fagnou, K. J. Am. Chem. Soc. 2004, 126, 9186–9187. doi:10.1021/ja049017y
Return to citation in text: [1] -
Campeau, L.-C.; Parisien, M.; Jean, A.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 581–590. doi:10.1021/ja055819x
Return to citation in text: [1] -
Liégault, B.; Petrov, I.; Gorelsky, S. I.; Fagnou, K. J. Org. Chem. 2010, 75, 1047–1060. doi:10.1021/jo902515z
Return to citation in text: [1] -
Paul, F.; Patt, J.; Hartwig, J. F. J. Am. Chem. Soc. 1994, 116, 5969–5970. doi:10.1021/ja00092a058
Return to citation in text: [1] -
Guram, A. S.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 7901–7902. doi:10.1021/ja00096a059
Return to citation in text: [1] -
Yamamoto, T.; Nishiyama, M.; Koie, Y. Tetrahedron Lett. 1998, 39, 2367–2370. doi:10.1016/s0040-4039(98)00202-0
Return to citation in text: [1] -
Dorel, R.; Grugel, C. P.; Haydl, A. M. Angew. Chem., Int. Ed. 2019, 58, 17118–17129. doi:10.1002/anie.201904795
Return to citation in text: [1] -
de Groot, L. H. M.; García‐Mateos, C.; Johnson, C. E.; Freyr Hlynsson, V.; Sharma, A. K.; Lomoth, R.; Wärnmark, K. Chem. – Eur. J. 2025, 31, e202500409. doi:10.1002/chem.202500409
Return to citation in text: [1] -
Brufani, G.; Bazzica, E.; Gu, Y.; Mauriello, F.; Vaccaro, L. Chin. Chem. Lett. 2026, 37, 111545. doi:10.1016/j.cclet.2025.111545
Return to citation in text: [1] -
Langer, P. Synlett 2024, 35, 1844–1860. doi:10.1055/s-0042-1751540
Return to citation in text: [1] -
Langer, P. Synlett 2022, 33, 1215–1226. doi:10.1055/s-0040-1719918
Return to citation in text: [1] -
Ustynyuk, N. A.; Lavrov, H. V.; Zarubin, D. N.; Dolgushin, F. M.; Ezernitskaya, M. G.; Gloriozov, I. P.; Zhokhov, S. S.; Zhokhova, N. I.; Ustynyuk, Y. A. Russ. Chem. Bull. 2018, 67, 1878–1890. doi:10.1007/s11172-018-2302-5
Return to citation in text: [1] [2] -
Asano, T.; Kobayashi, T.; Shimojo, K.; Yamaoka, S.; Torii, R.; Okano, K.; Yaita, T.; Mori, A. Preparation of multiply fused phenanthroline derivatives employing catalytic C-H arylation and the highly selective extraction of lanthanides. Presented at the 41st Symposium on Solvent Extraction, Tokyo, November 25, 2022.
Return to citation in text: [1] [2] -
Asano, T.; Nakanishi, Y.; Sugita, S.; Okano, K.; Narita, H.; Kobayashi, T.; Yaita, T.; Mori, A. ChemRxiv 2024. doi:10.26434/chemrxiv-2024-8lpmz
Return to citation in text: [1] [2] [3] -
Pramanik, S.; Li, B.; Driscoll, D. M.; Johnson, K. R.; Evans, B. R.; Damron, J. T.; Ivanov, A. S.; Jiang, D.-e.; Einkauf, J.; Popovs, I.; Jansone-Popova, S. J. Am. Chem. Soc. 2024, 146, 25669–25679. doi:10.1021/jacs.4c07332
Return to citation in text: [1] [2] -
Nakanishi, Y.; Sugita, S.; Okano, K.; Mori, A. Beilstein J. Org. Chem. 2024, 20, 3256–3262. doi:10.3762/bjoc.20.269
Return to citation in text: [1] [2] [3] -
Wang, S.; Lou, J.; Yang, X.; Xu, X.; Xu, L.; Fang, D.; Xu, C.; Xiao, C. Ind. Eng. Chem. Res. 2025, 64, 15830–15840. doi:10.1021/acs.iecr.5c01813
Return to citation in text: [1] [2] -
Hamada, Y.; Takeuchi, I.; Hirota, M. Chem. Pharm. Bull. 1974, 22, 485–492. doi:10.1248/cpb.22.485
Return to citation in text: [1] -
Chen, S.; Priebbenow, D. L.; Somkhit, J.; Scullino, C. V.; Agama, K.; Pommier, Y.; Flynn, B. L. Chem. – Eur. J. 2022, 28, e202201925. doi:10.1002/chem.202201925
Return to citation in text: [1] -
Yedage, S. L.; Bhanage, B. M. J. Org. Chem. 2016, 81, 4103–4111. doi:10.1021/acs.joc.6b00378
Return to citation in text: [1] -
Yu, Y.; Yue, Y.; Wang, D.; Li, X.; Chen, C.; Peng, J. Synthesis 2016, 48, 3941–3950. doi:10.1055/s-0035-1561481
Return to citation in text: [1] -
Saha, R.; Sekar, G. J. Catal. 2018, 366, 176–188. doi:10.1016/j.jcat.2018.08.009
Return to citation in text: [1] -
Manikandan, T. S.; Ramesh, R.; Semeril, D. Organometallics 2019, 38, 319–328. doi:10.1021/acs.organomet.8b00714
Return to citation in text: [1] -
Aleti, R. R.; Festa, A. A.; Voskressensky, L. G.; Van der Eycken, E. V. Molecules 2021, 26, 5560. doi:10.3390/molecules26185560
Return to citation in text: [1] -
Liu, Z.-S.; Xie, P.-P.; Hua, Y.; Wu, C.; Ma, Y.; Chen, J.; Cheng, H.-G.; Hong, X.; Zhou, Q. Chem 2021, 7, 1917–1932. doi:10.1016/j.chempr.2021.04.005
Return to citation in text: [1] -
Pan, C.; Wang, L.; Han, J. Adv. Synth. Catal. 2022, 364, 268–273. doi:10.1002/adsc.202100860
Return to citation in text: [1] -
Babu Pathi, V.; Manna, A.; Srinath, R.; Adhikary, S.; Banerji, B. Eur. J. Org. Chem. 2023, 26, e202300256. doi:10.1002/ejoc.202300256
Return to citation in text: [1] -
Li, L.-J.; Zhou, Z.-Q.; Liu, Z.-K.; He, Y.-Y.; Jia, F.-C.; Hu, X.-Q. Chem. Commun. 2023, 59, 438–441. doi:10.1039/d2cc05826j
Return to citation in text: [1] -
Jin, L.; Li, Y.; Mao, Y.; He, X.-B.; Lu, Z.; Zhang, Q.; Shi, B.-F. Nat. Commun. 2024, 15, 4908. doi:10.1038/s41467-024-48582-w
Return to citation in text: [1] -
Talukdar, V.; Paul, S.; Mondal, K.; Das, P. Adv. Synth. Catal. 2026, 368, e202401498. doi:10.1002/adsc.202401498
Return to citation in text: [1]
| 42. | Hamada, Y.; Takeuchi, I.; Hirota, M. Chem. Pharm. Bull. 1974, 22, 485–492. doi:10.1248/cpb.22.485 |
| 43. | Chen, S.; Priebbenow, D. L.; Somkhit, J.; Scullino, C. V.; Agama, K.; Pommier, Y.; Flynn, B. L. Chem. – Eur. J. 2022, 28, e202201925. doi:10.1002/chem.202201925 |
| 38. | Asano, T.; Nakanishi, Y.; Sugita, S.; Okano, K.; Narita, H.; Kobayashi, T.; Yaita, T.; Mori, A. ChemRxiv 2024. doi:10.26434/chemrxiv-2024-8lpmz |
| 40. | Nakanishi, Y.; Sugita, S.; Okano, K.; Mori, A. Beilstein J. Org. Chem. 2024, 20, 3256–3262. doi:10.3762/bjoc.20.269 |
| 36. | Ustynyuk, N. A.; Lavrov, H. V.; Zarubin, D. N.; Dolgushin, F. M.; Ezernitskaya, M. G.; Gloriozov, I. P.; Zhokhov, S. S.; Zhokhova, N. I.; Ustynyuk, Y. A. Russ. Chem. Bull. 2018, 67, 1878–1890. doi:10.1007/s11172-018-2302-5 |
| 37. | Asano, T.; Kobayashi, T.; Shimojo, K.; Yamaoka, S.; Torii, R.; Okano, K.; Yaita, T.; Mori, A. Preparation of multiply fused phenanthroline derivatives employing catalytic C-H arylation and the highly selective extraction of lanthanides. Presented at the 41st Symposium on Solvent Extraction, Tokyo, November 25, 2022. |
| 38. | Asano, T.; Nakanishi, Y.; Sugita, S.; Okano, K.; Narita, H.; Kobayashi, T.; Yaita, T.; Mori, A. ChemRxiv 2024. doi:10.26434/chemrxiv-2024-8lpmz |
| 39. | Pramanik, S.; Li, B.; Driscoll, D. M.; Johnson, K. R.; Evans, B. R.; Damron, J. T.; Ivanov, A. S.; Jiang, D.-e.; Einkauf, J.; Popovs, I.; Jansone-Popova, S. J. Am. Chem. Soc. 2024, 146, 25669–25679. doi:10.1021/jacs.4c07332 |
| 40. | Nakanishi, Y.; Sugita, S.; Okano, K.; Mori, A. Beilstein J. Org. Chem. 2024, 20, 3256–3262. doi:10.3762/bjoc.20.269 |
| 41. | Wang, S.; Lou, J.; Yang, X.; Xu, X.; Xu, L.; Fang, D.; Xu, C.; Xiao, C. Ind. Eng. Chem. Res. 2025, 64, 15830–15840. doi:10.1021/acs.iecr.5c01813 |
| 1. | Ganguly, A. K.; Wang, C. H.; David, M.; Bartner, P.; Chan, T. M. Tetrahedron Lett. 2002, 43, 6865–6868. doi:10.1016/s0040-4039(02)01537-x |
| 2. | Das, S.; Kundu, S.; Metya, A.; Maji, M. S. Chem. Sci. 2024, 15, 13466–13474. doi:10.1039/d4sc03438d |
| 8. | Pham, T. D. M.; Ziora, Z. M.; Blaskovich, M. A. T. Med. Chem. Commun. 2019, 10, 1719–1739. doi:10.1039/c9md00120d |
| 32. | de Groot, L. H. M.; García‐Mateos, C.; Johnson, C. E.; Freyr Hlynsson, V.; Sharma, A. K.; Lomoth, R.; Wärnmark, K. Chem. – Eur. J. 2025, 31, e202500409. doi:10.1002/chem.202500409 |
| 33. | Brufani, G.; Bazzica, E.; Gu, Y.; Mauriello, F.; Vaccaro, L. Chin. Chem. Lett. 2026, 37, 111545. doi:10.1016/j.cclet.2025.111545 |
| 34. | Langer, P. Synlett 2024, 35, 1844–1860. doi:10.1055/s-0042-1751540 |
| 35. | Langer, P. Synlett 2022, 33, 1215–1226. doi:10.1055/s-0040-1719918 |
| 5. | Michael, J. P. Nat. Prod. Rep. 2002, 19, 742–760. doi:10.1039/b104971m |
| 6. | Shang, X.-F.; Morris‐Natschke, S. L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Med. Res. Rev. 2018, 38, 775–828. doi:10.1002/med.21466 |
| 7. | Yadav, T. T.; Murahari, M.; Peters, G. J.; YC, M. Eur. J. Med. Chem. 2022, 239, 114527. doi:10.1016/j.ejmech.2022.114527 |
| 36. | Ustynyuk, N. A.; Lavrov, H. V.; Zarubin, D. N.; Dolgushin, F. M.; Ezernitskaya, M. G.; Gloriozov, I. P.; Zhokhov, S. S.; Zhokhova, N. I.; Ustynyuk, Y. A. Russ. Chem. Bull. 2018, 67, 1878–1890. doi:10.1007/s11172-018-2302-5 |
| 37. | Asano, T.; Kobayashi, T.; Shimojo, K.; Yamaoka, S.; Torii, R.; Okano, K.; Yaita, T.; Mori, A. Preparation of multiply fused phenanthroline derivatives employing catalytic C-H arylation and the highly selective extraction of lanthanides. Presented at the 41st Symposium on Solvent Extraction, Tokyo, November 25, 2022. |
| 38. | Asano, T.; Nakanishi, Y.; Sugita, S.; Okano, K.; Narita, H.; Kobayashi, T.; Yaita, T.; Mori, A. ChemRxiv 2024. doi:10.26434/chemrxiv-2024-8lpmz |
| 39. | Pramanik, S.; Li, B.; Driscoll, D. M.; Johnson, K. R.; Evans, B. R.; Damron, J. T.; Ivanov, A. S.; Jiang, D.-e.; Einkauf, J.; Popovs, I.; Jansone-Popova, S. J. Am. Chem. Soc. 2024, 146, 25669–25679. doi:10.1021/jacs.4c07332 |
| 40. | Nakanishi, Y.; Sugita, S.; Okano, K.; Mori, A. Beilstein J. Org. Chem. 2024, 20, 3256–3262. doi:10.3762/bjoc.20.269 |
| 41. | Wang, S.; Lou, J.; Yang, X.; Xu, X.; Xu, L.; Fang, D.; Xu, C.; Xiao, C. Ind. Eng. Chem. Res. 2025, 64, 15830–15840. doi:10.1021/acs.iecr.5c01813 |
| 5. | Michael, J. P. Nat. Prod. Rep. 2002, 19, 742–760. doi:10.1039/b104971m |
| 6. | Shang, X.-F.; Morris‐Natschke, S. L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Med. Res. Rev. 2018, 38, 775–828. doi:10.1002/med.21466 |
| 7. | Yadav, T. T.; Murahari, M.; Peters, G. J.; YC, M. Eur. J. Med. Chem. 2022, 239, 114527. doi:10.1016/j.ejmech.2022.114527 |
| 25. | Campeau, L.-C.; Parisien, M.; Leblanc, M.; Fagnou, K. J. Am. Chem. Soc. 2004, 126, 9186–9187. doi:10.1021/ja049017y |
| 26. | Campeau, L.-C.; Parisien, M.; Jean, A.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 581–590. doi:10.1021/ja055819x |
| 27. | Liégault, B.; Petrov, I.; Gorelsky, S. I.; Fagnou, K. J. Org. Chem. 2010, 75, 1047–1060. doi:10.1021/jo902515z |
| 3. | Guo, Z.; Yan, C.; Zhu, W.-H. Angew. Chem., Int. Ed. 2020, 59, 9812–9825. doi:10.1002/anie.201913249 |
| 4. | Yu, C. P.; Yamamoto, A.; Kumagai, S.; Takeya, J.; Okamoto, T. Angew. Chem., Int. Ed. 2023, 62, e202206417. doi:10.1002/anie.202206417 |
| 28. | Paul, F.; Patt, J.; Hartwig, J. F. J. Am. Chem. Soc. 1994, 116, 5969–5970. doi:10.1021/ja00092a058 |
| 29. | Guram, A. S.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 7901–7902. doi:10.1021/ja00096a059 |
| 30. | Yamamoto, T.; Nishiyama, M.; Koie, Y. Tetrahedron Lett. 1998, 39, 2367–2370. doi:10.1016/s0040-4039(98)00202-0 |
| 31. | Dorel, R.; Grugel, C. P.; Haydl, A. M. Angew. Chem., Int. Ed. 2019, 58, 17118–17129. doi:10.1002/anie.201904795 |
| 16. | King, A. O.; Okukado, N.; Negishi, E.-i. J. Chem. Soc., Chem. Commun. 1977, 683–684. doi:10.1039/c39770000683 |
| 17. | Muzammil; Zahoor, A. F.; Parveen, B.; Javed, S.; Akhtar, R.; Tabassum, S. Chem. Pap. 2024, 78, 3399–3430. doi:10.1007/s11696-024-03369-7 |
| 21. | Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866 |
| 22. | Lennox, A. J. J.; Lloyd-Jones, G. C. Chem. Soc. Rev. 2014, 43, 412–443. doi:10.1039/c3cs60197h |
| 14. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3 |
| 15. | Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084. doi:10.1039/c1cs15071e |
| 23. | Hatanaka, Y.; Hiyama, T. J. Org. Chem. 1988, 53, 918–920. doi:10.1021/jo00239a056 |
| 24. | Hiyama, T.; Minami, Y.; Mori, A. Transition-Metal-Catalyzed Cross-coupling of Organosilicon Compounds. In Organosilicon Chemistry; Hiyama, T.; Oestreich, M., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp 271–332. doi:10.1002/9783527814787.ch9 |
| 11. | Tamao, K.; Sumitani, K.; Kumada, M. J. Am. Chem. Soc. 1972, 94, 4374–4376. doi:10.1021/ja00767a075 |
| 12. | Corriu, R. J. P.; Masse, J. P. J. Chem. Soc., Chem. Commun. 1972, 144a. doi:10.1039/c3972000144a |
| 13. | Heravi, M. M.; Zadsirjan, V.; Hajiabbasi, P.; Hamidi, H. Monatsh. Chem. 2019, 150, 535–591. doi:10.1007/s00706-019-2364-6 |
| 44. | Yedage, S. L.; Bhanage, B. M. J. Org. Chem. 2016, 81, 4103–4111. doi:10.1021/acs.joc.6b00378 |
| 45. | Yu, Y.; Yue, Y.; Wang, D.; Li, X.; Chen, C.; Peng, J. Synthesis 2016, 48, 3941–3950. doi:10.1055/s-0035-1561481 |
| 46. | Saha, R.; Sekar, G. J. Catal. 2018, 366, 176–188. doi:10.1016/j.jcat.2018.08.009 |
| 47. | Manikandan, T. S.; Ramesh, R.; Semeril, D. Organometallics 2019, 38, 319–328. doi:10.1021/acs.organomet.8b00714 |
| 48. | Aleti, R. R.; Festa, A. A.; Voskressensky, L. G.; Van der Eycken, E. V. Molecules 2021, 26, 5560. doi:10.3390/molecules26185560 |
| 49. | Liu, Z.-S.; Xie, P.-P.; Hua, Y.; Wu, C.; Ma, Y.; Chen, J.; Cheng, H.-G.; Hong, X.; Zhou, Q. Chem 2021, 7, 1917–1932. doi:10.1016/j.chempr.2021.04.005 |
| 50. | Pan, C.; Wang, L.; Han, J. Adv. Synth. Catal. 2022, 364, 268–273. doi:10.1002/adsc.202100860 |
| 51. | Babu Pathi, V.; Manna, A.; Srinath, R.; Adhikary, S.; Banerji, B. Eur. J. Org. Chem. 2023, 26, e202300256. doi:10.1002/ejoc.202300256 |
| 52. | Li, L.-J.; Zhou, Z.-Q.; Liu, Z.-K.; He, Y.-Y.; Jia, F.-C.; Hu, X.-Q. Chem. Commun. 2023, 59, 438–441. doi:10.1039/d2cc05826j |
| 53. | Jin, L.; Li, Y.; Mao, Y.; He, X.-B.; Lu, Z.; Zhang, Q.; Shi, B.-F. Nat. Commun. 2024, 15, 4908. doi:10.1038/s41467-024-48582-w |
| 54. | Talukdar, V.; Paul, S.; Mondal, K.; Das, P. Adv. Synth. Catal. 2026, 368, e202401498. doi:10.1002/adsc.202401498 |
| 9. | Patra, S.; Hirose, T.; Himeda, Y. Inorg. Chim. Acta 2025, 585, 122769. doi:10.1016/j.ica.2025.122769 |
| 10. | Kumar, R.; Majumder, S.; Singh, C. P.; Samanta, C.; Newalkar, B. L.; Krishnamurty, S.; Das, R. K. ChemSusChem 2025, 18, e202502039. doi:10.1002/cssc.202502039 |
| 18. | Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Migita, T. Chem. Lett. 1977, 6, 301–302. doi:10.1246/cl.1977.301 |
| 19. | Milstein, D.; Stille, J. K. J. Am. Chem. Soc. 1978, 100, 3636–3638. doi:10.1021/ja00479a077 |
| 20. | Srivastav, N.; Singh, R.; Kaur, V. RSC Adv. 2015, 5, 62202–62213. doi:10.1039/c5ra09960a |
© 2026 Sugita et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.