Abstract
This review relates all the results that we obtained in the field of the total synthesis of indolizidine and quinolizidine alkaloids using a strategy of the addition of an allylsilane on an N-acyliminium ion. In this paper, we describe the synthesis of racemic indolizidine 167B and chiral indolizidines: (-)-indolizidines 167B, 195B, 223AB, (+)-monomorine, (-)-(3R,5S,8aS)-3-butyl-5-propylindolizidine and (-)-dendroprimine. Next, we relate the synthesis that we have developed in the quinolizidines field: (±)-myrtine and epimyrtine, (±)-lasubines I and II and chiral quinolizidines: (+)-myrtine, (-)-epimyrtine, (-)-lasubines I and II and (+)-subcosine II.

Graphical Abstract
Background
Bicyclic indolizidines and quinolizidines are commonly occurring structural skeleta found in natural alkaloids. Such compounds have been isolated from animals: poison frogs of the family Dendrobatidae have provided a rich source of novel pharmacologically active alkaloids, including a variety of bicyclic nitrogen heterocyclic compounds such as indolizidines. [1,2] Several quinolizidine alkaloids have been isolated from plants: Lythraceae family (Lasubines), [3]Vaccinum myrtillus (myrtine, epimyrtine). [4,5]
Firstly, most of these compounds are frequently found in concentrations too low to allow complete structural elucidation; secondly, the biological activities for most of them make these alkaloids ideal targets for total synthesis.
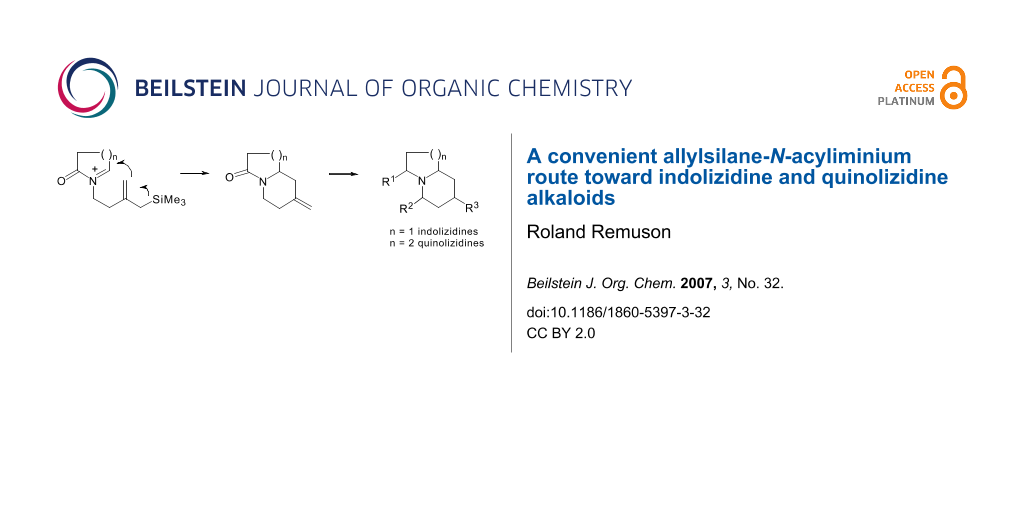
We have developed a new method to generate bicyclic indolizidine and quinolizidine compounds based on an intramolecular cyclisation of acyliminium ions substituted by an allylsilyl side chain as an internal π-nucleophile (Scheme 1). [6]
![[1860-5397-3-32-i1]](/bjoc/content/inline/1860-5397-3-32-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Allylsilane-N-acyliminium cyclisation.
Scheme 1: Allylsilane-N-acyliminium cyclisation.
This reaction has proven to be a very powerful method for construction of indolizidine and quinolizidine ring systems with efficient control of stereochemistry.
I Indolizidines
I 1. Indolizidine 167B
Indolizidine 167B, one of the simplest amphibian indolizidine alkaloids, was originally found as a trace component in the skin secretions of a frog belonging to the genus Dendrobates captured on the Isla Colon Panama. [7] The structure and relative stereochemistry shown in 1 are now accepted as correct although the absolute configuration of the natural product remains uncertain. [8] The lack of availability of the natural material and the important biological activities of the compound make this alkaloid an ideal target for total synthesis. [9-16]
I 1.1 Intramolecular cyclisation
We have found that intramolecular cyclisation of an allylsilane on an acyliminium ion constituted an excellent route to nitrogen bicyclic ring systems. [6] This method represents an efficient and stereoselective strategy for the preparation of 5-substituted indolizidines.
The source of chirality was the aminoester (R)-2 which was prepared according to Davies'methodology. [17] Synthesis of the indolizidine skeleton was carried out as shown in Scheme 2. Reaction of (R)-2 with succinic anhydride and then with acetyl chloride in refluxing toluene gave imide 3, then, 3 was reduced into ethoxylactam 4. In the next step, 4 was treated with two equivalents of the cerium reagent derived from trimethylsilymethylmagnesium chloride and CeCl3. The mixture was then hydrolysed with 1N HCl to give methylenindolizidinones 5a and 5b in a 4:1 ratio. Reduction of the mixture of lactams 5a and 5b with lithium aluminium hydride gave methylenindolizidines 6a and 6b which were separated by flash chromatography.
![[1860-5397-3-32-i2]](/bjoc/content/inline/1860-5397-3-32-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Enantioselective synthesis of (-)-indolizidine 167B by intramolecular allylsilane-N-acyliminium cyclisation.
Scheme 2: Enantioselective synthesis of (-)-indolizidine 167B by intramolecular allylsilane-N-acyliminium cyc...
Osmium tetroxide catalysed periodate oxidation of the olefinic bond of 6a and 6b led respectively to indolizidin-3-ones 7a and 7b. Upon treating an aqueous solution of 7a with 1N HCl the thermodynamically more stable indolizidinone 7b was obtained through a retro-Mannich fragmentation-cyclisation process. The last two steps were the conversion of 7b into its dithiolane and subsequent desulfurisation using Raney nickel. The synthesis of (-)-indolizidine 167B 1 has been achieved in 7 steps with a 17% overall yield from ethyl (3R)-3-aminohexanoate 2 with an enantiomeric excess of 93%. [19]
I 1.2 Intermolecular cyclisation
The intermolecular reaction between hydroxyalkyl-substituted allylsilanes and the acyliminium ion coming from pyrrolidin-2-one constitutes a new route to 5-substituted indolizidines (Scheme 3).
![[1860-5397-3-32-i3]](/bjoc/content/inline/1860-5397-3-32-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of (±)-indolizidine 167B by intermolecular cyclisation of allylsilane-N-acyliminium cyclisation.
Scheme 3: Synthesis of (±)-indolizidine 167B by intermolecular cyclisation of allylsilane-N-acyliminium cycli...
Hydroxyallylsilane 8 was synthesised as described [18] by reaction of the reagent prepared from allyltrimethylsilane, sec-butyllithium and titanium tetraisopropoxide with aldehydes. The key step of the synthesis is the intermolecular addition of the allylsilyl functional group of alcohol 8 on the acyliminium ion derived from ethoxycarbamate 9.
Treatment of a mixture of ethoxycarbamate 9 and hydroxyallylsilane 8 with one equivalent of stannic chloride resulted in the formation of 10 via the acyliminium ion intermediate 11. Subsequent oxidation of alcohol 10 with pyridinium dichromate, then catalytic hydrogenation (H2 over Pd/C) of ketone 12 induced hydrogenolysis of the CBz group, reduction of the double bond of the side chain and reduction of the iminium ion intermediate 13 to give the indolizidine 167B 1. [20]
The synthesis of (±)-indolizidine 167B has been achieved in five steps in 18% overall yield from pyrrolidin-2-one.
I 2. 3,5-Disubstituted indolizidines
Most of the indolizidine alkaloids are disubstituted by alkyl chains at the 3,5 positions. These compounds have been attractive targets for synthesis because of their potential biological activities. [7] Accordingly, novel strategies for the preparation of substituted indolizidines have received considerable attention. [21-27]
The allylsilyl functional group is a weak carbon nucleophile for trapping N-acyliminium ions, thus providing a useful method for intramolecular carbon-carbon bond formation. [28,29] We have applied this methodology towards the synthesis of indolizidine alkaloids. (vide supra) We describe here a new approach to 3,5-disubstituted indolizidines based on an intermolecular addition of allylsilanes on an N-acyl iminium starting from L-pyroglutamic acid used as the chiral precursor.
Preparation of lactam 14 was accomplished starting from the commercially available S-(-)-pyroglutamic acid according to a previously described procedure. [30,31] Next, lactam 14 was protected (n-BuLi, benzyl chloroformate) then converted to ethoxycarbamate 15, isolated as a mixture of two diastereomers according to Hiemstra's procedure. [32,33] Condensation of allylsilanes 16 onto iminium ion A generated in situ by treatment of 15 with stannous chloride led to compounds 17a and 17b. The next two steps were straightforward: oxidation (pyridinium dichromate) of 17a and 17b afforded α,β-ethylenic ketones 18a and 18b.
![[1860-5397-3-32-i4]](/bjoc/content/inline/1860-5397-3-32-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 3,5-disubstituted indolizidines from L-pyroglutamic acid.
Scheme 4: Synthesis of 3,5-disubstituted indolizidines from L-pyroglutamic acid.
On hydrogenation over palladium on carbon, 18a gave a mixture of indolizidines 19 and 20 which were separated by flash chromatography. They were identified as (-)-indolizidine 195B and (+)-monomorine respectively. In the same manner, the hydrogenation of 18b provided a mixture of isomers 21 and 22 respectively identified as (-)-indolizidine 223AB and (-)-(3R,5S,8aS)-3-butyl-5-propylindolizidine. [34] These four indolizidines were obtained in five steps with overall yields of about 8%.
I 3 (-)-Dendroprimine
(-)-Dendroprimine 22 is an alkaloid isolated from Dendrobium primulinum Lindl (Orchidaceae) and shown to be a 5,7-dimethylindolizidine. [35] Its relative configuration was determined after the synthesis of the four racemic diastereomers of this indolizidine and a conformational analysis of these diastereomers has been discussed. [36,37] Its identification as (5R,7S,9R)-5,7dimethylindolizidine has been firmly established. [38] We describe here the first asymmetric synthesis of this alkaloid; [39] two other syntheses were recently published. [40,41]
The first steps of our synthesis were carried out as shown in Scheme 5. The starting material was ethyl 2-aminopropanoate 23. Chirality was introduced with isomers (R)-23a and (S)-23b, which were prepared by a Michael reaction according to Davies' procedure from ethyl crotonate and respectively (R)- and (S)-N-benzyl-α-methylbenzylamine. [17] Reaction of 23a with succinic anhydride and then with acetyl chloride gave imide 24a, it was then reduced into ethoxylactam 25a. Compound 25a was treated with the cerium reagent derived from trimethylsilylmagnesium chloride and cerium chloride. The mixture was then hydrolysed with 1N HCl to give methylenindolizidinones 26a and 26b in a 4:1 ratio.
![[1860-5397-3-32-i5]](/bjoc/content/inline/1860-5397-3-32-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Access to indolizidine precursors of dendroprimine starting from chiral 2-aminopropanoate.
Scheme 5: Access to indolizidine precursors of dendroprimine starting from chiral 2-aminopropanoate.
These diastereomers could not be separated. According to Scheme 6, in the first step the reduction of the lactam functional group of cyclisation products 26a and 26b with lithium aluminium hydride afforded a 4:1 mixture of methylenindolizidines 27a and 27b in quantitative yield. These isomers were separated. Palladium-catalysed hydrogenation of 27a was found to be stereoselective, giving a mixture of 28a and (-)-(dendroprimine) 22 in a 3:1 ratio. Using similar conditions, 27b led to compound 29b.
![[1860-5397-3-32-i6]](/bjoc/content/inline/1860-5397-3-32-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Access to (-)-dendroprimine by reduction with LiAlH4 of indolizidinones 26.
Scheme 6: Access to (-)-dendroprimine by reduction with LiAlH4 of indolizidinones 26.
Another way (cf. Scheme 7) was studied to access (-)-dendroprimine 22: hydrogenation of the crude mixture of cyclisation products 26a and 26b over palladium on carbon provided a mixture of lactams 29a, 30a and 31a in which isomer 30a was preponderant (29a/30a/31a = 15:65:20). Flash column chromatography gave pure 31a in 18% yield, but 29a and 30a could not be separated (50% yield). A mixture of the three isomers was used without purification for the next step. This mixture was then reduced with lithium aluminium hydride to give the indolizidines 28a, 22 and 29b. In conclusion, (-)-dendroprimine was obtained in five steps with overall yields of 17 and 20%.
![[1860-5397-3-32-i7]](/bjoc/content/inline/1860-5397-3-32-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Access to (-)-dendroprimine by catalytic hydrogenation of indolizidinones 26.
Scheme 7: Access to (-)-dendroprimine by catalytic hydrogenation of indolizidinones 26.
II Quinolizidines
II 1-Myrtine and epimyrtine
(+)-Myrtine and (-)-epimyrtine are quinolizidine alkaloids isolated from Vaccinium myrtillus (Ericaceae). [4,5] Several syntheses of these compounds as racemic mixtures have been described, [5,42-44] but only three enantioselective syntheses of (+)-myrtine [43,45] and three syntheses of (-)-epimyrtine have been published. [46,47]
II 1.1 Synthesis of (±)-myrtine and (±)-epimyrtine
These compounds have been prepared according to Scheme 8, the synthesis of hydroxyalkylallylsilane 32 is accomplished in 40% yield following Trost's procedure. [48] Reaction of glutarimide with alcohol 32 under Mitsunobu reaction conditions afforded imide 33 in 67% yield. Reduction of 33 was carried out with an excess of sodium borohydride in methanol at 0°C to give 34 as a mixture of two diastereomers which were not separated. The hydroxylactam 34 was then cyclised to the quinolizidine isomers 35a and 35b on treatment with 4 equiv. of trifluoroacetic acid in a 7:3 ratio. Then, ozonolysis of 35a and 35b followed by reduction with dimethylsulfide furnished respectively 36a and 36b. Protection of the carbonyl group by ketalisation with 2-ethyl-2-methyl-1,3-dioxolane and p-toluenesulfonic acid, reduction of the amide function with lithium aluminium hydride then quantitative removal of the protecting group (HCl treatment) afforded (±)-myrtine and (±)-epimyrtine. These syntheses were achieved in seven steps and 20% overall yield. [49]
![[1860-5397-3-32-i8]](/bjoc/content/inline/1860-5397-3-32-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of (±)-myrtine and (±)-epimyrtine.
Scheme 8: Synthesis of (±)-myrtine and (±)-epimyrtine.
II 1.2 Synthesis of (+)-myrtine and (-)-epimyrtine
We used a similar strategy to prepare the enantiopure compounds starting from (S)-2-(hydroxypropyl)allyltrimethylsilane 32 (cf. Scheme 9). Compound 32 was obtained in quantitative yield by cerium mediated trimethylsilylmethylmagnesium chloride addition to ethyl (S)-3-hydroxybutanoate as we described. [50] The first three steps of the enantioselective synthesis were those previously described for the synthesis of racemic compounds (vide supra). Condensation of alcohol 32 with glutarimide under Mitsunobu conditions led to (+)-imide (R)-33 in 67% yield. Reduction of (R)-33 with sodium borohydride afforded hydroxylactam 37 as a 1:1 mixture of isomers in 95% yield. Treatment of hydroxylactam 37 with trifluoroacetic acid in methylene chloride gave a 7:3 mixture of the two isomeric bicyclic compounds (4R,10R)-35a and (4R,10S)-35b in quantitative yield. Reduction of this mixture of lactams with lithium aluminium hydride gave a 7:3 mixture of methylenquinolizidines 38a and 38b in quantitative yield. Osmium tetroxide-catalysed periodate oxidation of the olefinic bond of quinolizidines 38a and 38b under carefully controlled conditions led to a 7:3 mixture of the two diastereomeric alkaloids (+)-myrtine and (-)-epimyrtine. These alkaloids were obtained in five steps from (S)-2-(2-hydroxypropyl)allylsilane 32 with an overall yield of 23% and a high enantiomeric purity. This synthesis constitutes the first total synthesis of naturally occurring (-)-epimyrtine and confirms the configuration 4R,10S which was assigned previously to this compound. [51]
![[1860-5397-3-32-i9]](/bjoc/content/inline/1860-5397-3-32-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Enantioselective synthesis of (+)-myrtine and (-)-epimyrtine.
Scheme 9: Enantioselective synthesis of (+)-myrtine and (-)-epimyrtine.
II 2. Lasubines
The Lythraceae alkaloids constitute a large family of natural products, most of which contain 4-arylquinolizidine substructures. Among them are the quinolizidine alkaloids lasubine I and lasubine II which have been isolated from Lagerstroemia subscotata Koehne. [3] Numerous racemic [44,52-54] and asymmetric total syntheses of these alkaloids have been described. [43,55-64]
II 2.1. Synthesis of (±)-lasubine I and (±)-lasubine II
The first steps of our synthesis were carried out as shown in Scheme 10. The starting material was 2-(2-hydroxyethyl)allylsilane 39 which was prepared in 86% yield by indium mediated allylsilylation of 3,4-dimethoxybenzaldehyde, as already described. [65] Condensation of alcohol 39 with glutarimide under Mitsunobu conditions led to imide 40 in 46% yield. Reduction of 40 with diisobutylaluminium hydride afforded hydroxylactam 41 isolated as a mixture of isomers, a higher yield of a single isomer was obtained when using lithium triethylborohydride as reducing regent. The reduction had to be performed at -78°C to prevent formation of ring opening products. [66] Treatment of hydroxylactam 41 with trifluoroacetic acid in methylene chloride gave a mixture of isomeric bicyclic compounds 42a and 42b in a quantitative yield and a 4:1 ratio when the reaction was performed at -78°C.
![[1860-5397-3-32-i10]](/bjoc/content/inline/1860-5397-3-32-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of (±)-lasubines I and II and (±)-2-epilasubine II.
Scheme 10: Synthesis of (±)-lasubines I and II and (±)-2-epilasubine II.
Then, we examined two routes to the quinolizidine alkaloids lasubine I and lasubine II from methylenquinolizidinones 42a and 42b. They involved oxidation of the methylene group into a carbonyl which was then stereoselectively reduced to the hydroxyl group. The shortest route consisted of the ozonolysis of the methylene group followed by the simultaneous reduction of the two carbonyl groups of keto lactams 43a and 43b. Thus, treatment of 42a with ozone then with dimethyl sulfide afforded the expected keto lactam 43a in 91% yield. Ozonolysis of 42b led to keto lactam 43b in 63% yield. Reduction of 43a with lithium aluminium hydride afforded in 60% yield a 1:1.2 mixture of lasubine I and 2-epilasubine I which were separated as their acetates. In the same way, reduction of 43b gave 2-epilasubine II in 70% yield.
In order to circumvent the stereochemical difficulty we decided to reduce first the lactam group (cf. Scheme 11) to obtain quinolizidines whose conformation should not be distorted by the junction with the piperidone ring. Lactams 42a and 42b were reduced with lithium aluminium hydride to give methylenquinolizidines 44a and 44b in 92% and 78% yields respectively. Osmium tetroxide catalysed periodate oxidation of the olefinic bond of methylenquinolizidines 44a and 44b under carefully controlled conditions led to the already described 2-oxoquinolizidines 45a and 45b in 79% and 89% yields respectively. The final step is a reduction of the carbonyl group. The use of borohydride in the reduction of 45a has been described to give lasubine in an excellent yield. [67,68] In our hands, this reaction afforded a 1:1 mixture of (±)-lasubine I and (±)-epilasubine I. Stereoselective reduction of quinolizidin-2-one 45a to (±)-lasubine I was achieved in 50% yield with lithium tri-sec-butylborohydride (L-selectride). Quinolizidinone 45b was selectively converted to (±)-lasubine II with lithium trisamylborohydride (LS-selectride) in 60% yield.
![[1860-5397-3-32-i11]](/bjoc/content/inline/1860-5397-3-32-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of (±)-lasubine I and II.
Scheme 11: Synthesis of (±)-lasubine I and II.
In conclusion, (±) lasubine I and (±)-lasubine II were obtained in six steps from 2-(2-hydroxyethyl)allylsilane 39 in 8% and 7.4% yields respectively.
II.2.2 Synthesis of (-)-lasubine I, (-)-lasubine II and (+)-subcosine II
A similar strategy was attempted from (+)-(3R)-ethyl 3-hydroxy-3-(3,4-dimethoxyphenyl)propionate but racemisation was observed during the Mitsunobu reaction. [69] So we developed another strategy to prepare these natural optically active compounds based on the intramolecular acyliminium ion allylsilane cyclisation of intermediate 49 generated from ethoxylactam 48. Chirality is introduced with the β-aminoester 46.
(S)-β-Aminoester 46 was prepared according to Davies'procedure. [17] Reaction of 46 with glutaric anhydride then with acetyl chloride in refluxing toluene gave imide 47 in 86% yield. Imide 47 was reduced into ethoxylactam 48 which was isolated as a mixture of two diastereomers. In the next step, ethoxylactam 48 was treated with the cerium reagent derived from CeCl3 and trimethylsilylmethylmagnesium chloride. The mixture was then hydrolysed with 1N HCl to give methylenquinolizidinones 42a and 42b in a 1:5 ratio and 60% yield. Reduction of lactams 42a and 42b with lithium aluminium hydride in refluxing THF for 12 h gave methylenquinolizidines 44a and 44b in 83% and 92% yields respectively. Osmium tetroxide catalysed periodate oxidation of the olefinic bond of 44a and 44b under carefully controlled conditions led to quinolizidin-2-ones 45a and 45b in 70 and 90% yields. The final step is a reduction of the carbonyl group. Stereoselective reduction of 45a with L-selectride provided (-)-lasubine I in 62% yield. Quinolizidin-2-one 45b was selectively converted to (-)-lasubine II with LS-selectride in 65% yield. Acylation of (-)-lasubine II with 3,4-dimethoxycinnamic anhydride gave (+)-subcosine II in 60% yield.(Scheme 12)
![[1860-5397-3-32-i12]](/bjoc/content/inline/1860-5397-3-32-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Enantioselective synthesis of (-)-lasubines I and II and (+)-subcosine.
Scheme 12: Enantioselective synthesis of (-)-lasubines I and II and (+)-subcosine.
In conclusion, we have described the total synthesis of (-)-lasubine I, (-)-lasubine II and (+)-subcosine II using intramolecular cyclisation of N-acyliminium ion (S)-49. (-)-Lasubine I and (-)-lasubine II were obtained in six steps with overall yields of 7 and 14% respectively. (+)-Subcosine was prepared in seven steps with an overall yield of 9%. These three compounds were obtained with high enantiomeric purity. These results constitute the first total synthesis of naturally occurring (-)-lasubine II and (+)-subcosine II and unambiguously establish their absolute configuration as 2S,4S,10S.
References
-
Daly, J. W.; Spande, T. F. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; New York: Wiley Interscience, 1986; Vol. 4, pp 1–274.
Return to citation in text: [1] -
Tokuyama, T.; Nishiromi, N.; Karle, I. L.; Ewars, M. W.; Daly, J. W. Tetrahedron 1986, 42, 3453–3460. doi:10.1016/S0040-4020(01)87312-9
Return to citation in text: [1] -
Fuji, K.; Yamada, T.; Fujita, E.; Murata, H. Chem. Pharm. Bull. 1978, 26, 2515–2521.
Return to citation in text: [1] [2] -
Slosse, P.; Hootelé, C. Tetrahedron Lett. 1978, 397–398. doi:10.1016/S0040-4039(01)85135-2
Return to citation in text: [1] [2] -
Slosse, P.; Hootelé, C. Tetrahedron 1981, 37, 4287–4294. doi:10.1016/0040-4020(81)85024-7
Return to citation in text: [1] [2] [3] -
Gelas-Mialhe, Y.; Gramain, J. C.; Hajouji, H.; Remuson, R. Heterocycles 1992, 34, 37–49.
Return to citation in text: [1] [2] -
Daly, J. W.; Garrafo, H. M.; Spande, T. F. Alkaloids; New York: Wiley Interscience, 1993; Vol. 43, pp 185–288.
Return to citation in text: [1] [2] -
Michael, J. P.; Gravestock, D. Eur. J. Org. Chem. 1998, 865–870. doi:10.1002/(SICI)1099-0690(199805)1998:5<865::AID-EJOC865>3.0.CO;2-3
Return to citation in text: [1] -
Guazzelli, G.; Lazzaroni, R.; Settambolo, R. Synthesis 2005, 3119–3123.
Return to citation in text: [1] -
Roa, L. F.; Gnecco, D.; Galindo, A.; Teran, J. L. Tetrahedron: Asymmetry 2004, 15, 3393–3395. doi:10.1016/j.tetasy.2004.09.030
Return to citation in text: [1] -
Reddy, P. G.; Baskaran, S. J. Org. Chem. 2004, 69, 3093–3101. doi:10.1021/jo035258x
Return to citation in text: [1] -
Amat, M.; Llor, N.; Hidalgo, J.; Escolano, C.; Bosch, J. J. Org. Chem. 2003, 68, 1919–1928. doi:10.1021/jo0266083
Return to citation in text: [1] -
Reddy, P. G.; Varghese, B.; Baskaran, S. Org. Lett. 2003, 5, 583–585. doi:10.1021/ol027563v
Return to citation in text: [1] -
Carbonnel, S.; Troin, Y. Heterocycles 2002, 57, 1807–1830.
Return to citation in text: [1] -
Zaminer, J.; Stapper, C.; Blechert, S. Tetrahedron Lett. 2002, 43, 6739–6741. doi:10.1016/S0040-4039(02)01534-4
Return to citation in text: [1] -
Corvo, M.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X
Return to citation in text: [1] -
Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X
Return to citation in text: [1] [2] [3] -
Reetz, M. T.; Steinbach, R.; Westermann, J.; Meter, R.; Wenderoth, B. Chem. Ber. 1985, 118, 1441–1454. doi:10.1002/cber.19851180413
Return to citation in text: [1] -
Chalard, P.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C.; Canet, I. Tetrahedron Lett. 1999, 40, 1661–1664. doi:10.1016/S0040-4039(99)00059-3
Return to citation in text: [1] -
Peroche, S.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Tetrahedron Lett. 2001, 42, 4617–4619. doi:10.1016/S0040-4039(01)00822-X
Return to citation in text: [1] -
Toyooka, N.; Nemoto, H. Heterocycles 2005, 66, 549–555.
Return to citation in text: [1] -
Smith, A. B., III; Kim, D.-S. J. Org. Chem. 2006, 71, 2547–2557. doi:10.1021/jo052314g
Return to citation in text: [1] -
Smith, A. B., III; Kim, D.-S. Org. Lett. 2004, 6, 1493–1495. doi:10.1021/ol049601b
Return to citation in text: [1] -
Kiewel, K. J.; Tallant, M.; Sulikowski, G. A. Tetrahedron Lett. 2001, 42, 6621–6623. doi:10.1016/S0040-4039(01)01361-2
Return to citation in text: [1] -
Lee, E.; Jeong, E.; Min, S. J.; Hong, S.; Lim, J.; Kim, S. K.; Sang, K.; Kim, H. J.; Choi, B. G.; Koo, K. C. Org. Lett. 2000, 2, 2169–2171. doi:10.1021/ol006094z
Return to citation in text: [1] -
Celimene, C.; Dhimane, H.; Lhommet, G. Tetrahedron 1998, 54, 10457–10468. doi:10.1016/S0040-4020(98)00498-0
Return to citation in text: [1] -
Mori, M.; Hori, M.; Sato, Y. J. Org. Chem. 1998, 63, 4832–4833. doi:10.1021/jo980288z
Return to citation in text: [1] -
Hiemstra, H.; Fortgens, H. P.; Speckamp, W. N. Tetrahedron Lett. 1985, 26, 3155–3158. doi:10.1016/S0040-4039(00)98644-1
Return to citation in text: [1] -
Agami, C.; Comesse, S.; Kadouri-Puchot, C. J. Org. Chem. 2000, 65, 4435–4439. doi:10.1021/jo0000271
Return to citation in text: [1] -
Pilli, R. A.; Dias, C.; Maldaner, A. O. J. Org. Chem. 1995, 60, 717–722. doi:10.1021/jo00108a040
references cited therein.
Return to citation in text: [1] -
Saliou, C.; Fleurant, A.; Célérier, J. P.; Lhommet, G. Tetrahedron Lett. 1991, 32, 3365–3368. doi:10.1016/S0040-4039(00)92707-2
Return to citation in text: [1] -
Malching, K. H.; Hiemstra, H.; Klaver, W. J.; Speckamp, W. N. Tetrahedron Lett. 1986, 27, 4799–4802. doi:10.1016/S0040-4039(00)85068-6
Return to citation in text: [1] -
Klaver, W. J.; Hiemstra, H.; Speckamp, W. N. Tetrahedron Lett. 1987, 28, 1581–1584. doi:10.1016/S0040-4039(01)81047-9
Return to citation in text: [1] -
Conchon, E.; Gelas-Mialhe, Y.; Remuson, R. Tetrahedron: Asymmetry 2006, 17, 1253–1257. doi:10.1016/j.tetasy.2006.04.027
Return to citation in text: [1] -
Lüning, B.; Leander, K. Acta Chem. Scand. 1965, 19, 1607–1611.
Return to citation in text: [1] -
Lüning, B.; Lundin, C. Acta Chem. Scand. 1967, 21, 2136–2142.
Return to citation in text: [1] -
Sonnet, P. E.; Netzel, D. A.; Mendoza, R. J. Heterocycl. Chem. 1979, 16, 1041–1047.
Return to citation in text: [1] -
Blomquist, L.; Leander, K.; Lüning, B.; Rosemblom, J. Acta Chem. Scand. 1972, 26, 3203–3206.
Return to citation in text: [1] -
De Saboulin Bollena, A.; Gelas-Mialhe, Y.; Gramain, J. C.; Perret, A.; Remuson, R. J. Nat. Prod. 2004, 67, 1029–1031. doi:10.1021/np049902u
Return to citation in text: [1] -
Kobayashi, T.; Hasegawa, F.; Katsunori, T.; Katsumura, S. Org. Lett. 2006, 8, 5917–5921. doi:10.1021/ol0622716
Return to citation in text: [1] -
Kobayashi, T.; Hasegawa, F.; Tanaka, K.; Katsumura, S. Org. Lett. 2006, 8, 3813–3816. doi:10.1021/ol0614065
Return to citation in text: [1] -
King, F. D. J. Chem. Soc., Perkin Trans. 1 1986, 447–453. doi:10.1039/p19860000447
Return to citation in text: [1] -
Comins, D. L.; LaMunyon, D. H. J. Org. Chem. 1992, 57, 5807–5809. doi:10.1021/jo00048a003
Return to citation in text: [1] [2] [3] -
Pilli, R. A.; Dias, L. C.; Maldaner, A. O. J. Org. Chem. 1995, 60, 717–722. doi:10.1021/jo00108a040
Return to citation in text: [1] [2] -
Davis, F. A.; Xu, H.; Zhang, J. J. Org. Chem. 2007, 72, 2046–2052. doi:10.1021/jo062365t
Return to citation in text: [1] -
Amore, S. M.; Judd, A. S.; Martin, S. F. Org. Lett. 2005, 7, 2031–2033. doi:10.1021/ol050544b
Return to citation in text: [1] -
Davis, F. A.; Zhang, Y.; Anilkumar, G. J. Org. Chem. 2003, 68, 8061–8064. doi:10.1021/jo030208d
Return to citation in text: [1] -
Trost, B. M.; Chan, D. M. T.; Nanninga, T. N. Org. Synth. 1984, 62, 58.
Return to citation in text: [1] -
Gelas-Mialhe, Y.; Gramain, J. C.; Louvet, A.; Remuson, R. Tetrahedron Lett. 1992, 33, 73–76. doi:10.1016/S0040-4039(00)77676-3
Return to citation in text: [1] -
Bardot, V.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Tetrahedron: Asymmetry 1997, 8, 1111–1114. doi:10.1016/S0957-4166(97)00079-7
Return to citation in text: [1] -
Gardette, D.; Gelas-Mialhe, Y.; Gramain, J. C.; Perrin, B.; Remuson, R. Tetrahedron: Asymmetry 1998, 9, 1823–1838. doi:10.1016/S0957-4166(98)00170-0
Return to citation in text: [1] -
Pilli, R. A.; Dias, L. C.; Maldaner, A. O. Tetrahedron Lett. 1993, 34, 2729–2732. doi:10.1016/S0040-4039(00)73547-7
Return to citation in text: [1] -
Brown, J. D.; Foley, M. A.; Comins, D. L. J. Am. Chem. Soc. 1988, 110, 7445–7447. doi:10.1021/ja00230a026
Return to citation in text: [1] -
Ent, H.; De Koning, H.; Speckamp, W. N. Heterocycles 1988, 27, 237–243.
Return to citation in text: [1] -
Liu, S.; Fan, Y.; Peng, X.; Wang, W.; Hua, W.; Akber, H.; Liao, L. Tetrahedron Lett. 2006, 47, 7681–7684. doi:10.1016/j.tetlet.2006.08.137
Return to citation in text: [1] -
Yu, R. T.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 12370–12371. doi:10.1021/ja064868m
Return to citation in text: [1] -
Back, T. G.; Hamilton, M. D.; Lim, V. J. J.; Parvez, M. J. Org. Chem. 2005, 70, 967–972. doi:10.1021/jo048284j
Return to citation in text: [1] -
Zaja, M.; Blechert, S. Tetrahedron 2004, 60, 9629–9634. doi:10.1016/j.tet.2004.06.145
Return to citation in text: [1] -
Gracias, V.; Zeng, Y.; Desai, P.; Aube, J. Org. Lett. 2003, 5, 4999–5001. doi:10.1021/ol035965c
Return to citation in text: [1] -
Back, T. G.; Hamilton, M. D. Org. Lett. 2002, 4, 1779–1781. doi:10.1021/ol0258482
Return to citation in text: [1] -
Ma, D.; Zhu, W. Org. Lett. 2001, 3, 3927–3929. doi:10.1021/ol016802w
Return to citation in text: [1] -
Davis, F. A.; Chao, B. Org. Lett. 2000, 2, 2623–2625. doi:10.1021/ol0061438
Return to citation in text: [1] -
Ratni, H.; Kündig, E. P. Org. Lett. 2000, 2, 1983. doi:10.1021/ol006092e
Return to citation in text: [1] -
Ratni, H.; Kündig, E. P. Org. Lett. 1999, 1, 1997–1999. doi:10.1021/ol991158v
Return to citation in text: [1] -
Bardot, V.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Synlett 1996, 37–38. doi:10.1055/s-1996-5310
Return to citation in text: [1] -
Hubert, J. C.; Wijnberg, J. B. P. A.; Speckamp, W. N. Tetrahedron 1975, 31, 1437–1441.
Return to citation in text: [1] -
Iiada, H.; Tanaka, M.; Kibayashi, C. J. Org. Chem. 1984, 49, 1909–1911. doi:10.1021/jo00185a012
Return to citation in text: [1] -
Beckwith, A. L.; Joseph, S. P.; Mayadunne, T. A. J. Org. Chem. 1993, 58, 4198–4199. doi:10.1021/jo00068a009
Return to citation in text: [1] -
Bardot, V. Ph.D. Thesis, Université Clermont-Ferrand, Clermont-Ferrand, France, 1994.
Return to citation in text: [1]
| 46. | Amore, S. M.; Judd, A. S.; Martin, S. F. Org. Lett. 2005, 7, 2031–2033. doi:10.1021/ol050544b |
| 47. | Davis, F. A.; Zhang, Y.; Anilkumar, G. J. Org. Chem. 2003, 68, 8061–8064. doi:10.1021/jo030208d |
| 49. | Gelas-Mialhe, Y.; Gramain, J. C.; Louvet, A.; Remuson, R. Tetrahedron Lett. 1992, 33, 73–76. doi:10.1016/S0040-4039(00)77676-3 |
| 1. | Daly, J. W.; Spande, T. F. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; New York: Wiley Interscience, 1986; Vol. 4, pp 1–274. |
| 2. | Tokuyama, T.; Nishiromi, N.; Karle, I. L.; Ewars, M. W.; Daly, J. W. Tetrahedron 1986, 42, 3453–3460. doi:10.1016/S0040-4020(01)87312-9 |
| 7. | Daly, J. W.; Garrafo, H. M.; Spande, T. F. Alkaloids; New York: Wiley Interscience, 1993; Vol. 43, pp 185–288. |
| 28. | Hiemstra, H.; Fortgens, H. P.; Speckamp, W. N. Tetrahedron Lett. 1985, 26, 3155–3158. doi:10.1016/S0040-4039(00)98644-1 |
| 29. | Agami, C.; Comesse, S.; Kadouri-Puchot, C. J. Org. Chem. 2000, 65, 4435–4439. doi:10.1021/jo0000271 |
| 66. | Hubert, J. C.; Wijnberg, J. B. P. A.; Speckamp, W. N. Tetrahedron 1975, 31, 1437–1441. |
| 6. | Gelas-Mialhe, Y.; Gramain, J. C.; Hajouji, H.; Remuson, R. Heterocycles 1992, 34, 37–49. |
| 30. |
Pilli, R. A.; Dias, C.; Maldaner, A. O. J. Org. Chem. 1995, 60, 717–722. doi:10.1021/jo00108a040
references cited therein. |
| 31. | Saliou, C.; Fleurant, A.; Célérier, J. P.; Lhommet, G. Tetrahedron Lett. 1991, 32, 3365–3368. doi:10.1016/S0040-4039(00)92707-2 |
| 67. | Iiada, H.; Tanaka, M.; Kibayashi, C. J. Org. Chem. 1984, 49, 1909–1911. doi:10.1021/jo00185a012 |
| 68. | Beckwith, A. L.; Joseph, S. P.; Mayadunne, T. A. J. Org. Chem. 1993, 58, 4198–4199. doi:10.1021/jo00068a009 |
| 4. | Slosse, P.; Hootelé, C. Tetrahedron Lett. 1978, 397–398. doi:10.1016/S0040-4039(01)85135-2 |
| 5. | Slosse, P.; Hootelé, C. Tetrahedron 1981, 37, 4287–4294. doi:10.1016/0040-4020(81)85024-7 |
| 7. | Daly, J. W.; Garrafo, H. M.; Spande, T. F. Alkaloids; New York: Wiley Interscience, 1993; Vol. 43, pp 185–288. |
| 43. | Comins, D. L.; LaMunyon, D. H. J. Org. Chem. 1992, 57, 5807–5809. doi:10.1021/jo00048a003 |
| 55. | Liu, S.; Fan, Y.; Peng, X.; Wang, W.; Hua, W.; Akber, H.; Liao, L. Tetrahedron Lett. 2006, 47, 7681–7684. doi:10.1016/j.tetlet.2006.08.137 |
| 56. | Yu, R. T.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 12370–12371. doi:10.1021/ja064868m |
| 57. | Back, T. G.; Hamilton, M. D.; Lim, V. J. J.; Parvez, M. J. Org. Chem. 2005, 70, 967–972. doi:10.1021/jo048284j |
| 58. | Zaja, M.; Blechert, S. Tetrahedron 2004, 60, 9629–9634. doi:10.1016/j.tet.2004.06.145 |
| 59. | Gracias, V.; Zeng, Y.; Desai, P.; Aube, J. Org. Lett. 2003, 5, 4999–5001. doi:10.1021/ol035965c |
| 60. | Back, T. G.; Hamilton, M. D. Org. Lett. 2002, 4, 1779–1781. doi:10.1021/ol0258482 |
| 61. | Ma, D.; Zhu, W. Org. Lett. 2001, 3, 3927–3929. doi:10.1021/ol016802w |
| 62. | Davis, F. A.; Chao, B. Org. Lett. 2000, 2, 2623–2625. doi:10.1021/ol0061438 |
| 63. | Ratni, H.; Kündig, E. P. Org. Lett. 2000, 2, 1983. doi:10.1021/ol006092e |
| 64. | Ratni, H.; Kündig, E. P. Org. Lett. 1999, 1, 1997–1999. doi:10.1021/ol991158v |
| 3. | Fuji, K.; Yamada, T.; Fujita, E.; Murata, H. Chem. Pharm. Bull. 1978, 26, 2515–2521. |
| 21. | Toyooka, N.; Nemoto, H. Heterocycles 2005, 66, 549–555. |
| 22. | Smith, A. B., III; Kim, D.-S. J. Org. Chem. 2006, 71, 2547–2557. doi:10.1021/jo052314g |
| 23. | Smith, A. B., III; Kim, D.-S. Org. Lett. 2004, 6, 1493–1495. doi:10.1021/ol049601b |
| 24. | Kiewel, K. J.; Tallant, M.; Sulikowski, G. A. Tetrahedron Lett. 2001, 42, 6621–6623. doi:10.1016/S0040-4039(01)01361-2 |
| 25. | Lee, E.; Jeong, E.; Min, S. J.; Hong, S.; Lim, J.; Kim, S. K.; Sang, K.; Kim, H. J.; Choi, B. G.; Koo, K. C. Org. Lett. 2000, 2, 2169–2171. doi:10.1021/ol006094z |
| 26. | Celimene, C.; Dhimane, H.; Lhommet, G. Tetrahedron 1998, 54, 10457–10468. doi:10.1016/S0040-4020(98)00498-0 |
| 27. | Mori, M.; Hori, M.; Sato, Y. J. Org. Chem. 1998, 63, 4832–4833. doi:10.1021/jo980288z |
| 65. | Bardot, V.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Synlett 1996, 37–38. doi:10.1055/s-1996-5310 |
| 17. | Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X |
| 18. | Reetz, M. T.; Steinbach, R.; Westermann, J.; Meter, R.; Wenderoth, B. Chem. Ber. 1985, 118, 1441–1454. doi:10.1002/cber.19851180413 |
| 3. | Fuji, K.; Yamada, T.; Fujita, E.; Murata, H. Chem. Pharm. Bull. 1978, 26, 2515–2521. |
| 6. | Gelas-Mialhe, Y.; Gramain, J. C.; Hajouji, H.; Remuson, R. Heterocycles 1992, 34, 37–49. |
| 20. | Peroche, S.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Tetrahedron Lett. 2001, 42, 4617–4619. doi:10.1016/S0040-4039(01)00822-X |
| 44. | Pilli, R. A.; Dias, L. C.; Maldaner, A. O. J. Org. Chem. 1995, 60, 717–722. doi:10.1021/jo00108a040 |
| 52. | Pilli, R. A.; Dias, L. C.; Maldaner, A. O. Tetrahedron Lett. 1993, 34, 2729–2732. doi:10.1016/S0040-4039(00)73547-7 |
| 53. | Brown, J. D.; Foley, M. A.; Comins, D. L. J. Am. Chem. Soc. 1988, 110, 7445–7447. doi:10.1021/ja00230a026 |
| 54. | Ent, H.; De Koning, H.; Speckamp, W. N. Heterocycles 1988, 27, 237–243. |
| 9. | Guazzelli, G.; Lazzaroni, R.; Settambolo, R. Synthesis 2005, 3119–3123. |
| 10. | Roa, L. F.; Gnecco, D.; Galindo, A.; Teran, J. L. Tetrahedron: Asymmetry 2004, 15, 3393–3395. doi:10.1016/j.tetasy.2004.09.030 |
| 11. | Reddy, P. G.; Baskaran, S. J. Org. Chem. 2004, 69, 3093–3101. doi:10.1021/jo035258x |
| 12. | Amat, M.; Llor, N.; Hidalgo, J.; Escolano, C.; Bosch, J. J. Org. Chem. 2003, 68, 1919–1928. doi:10.1021/jo0266083 |
| 13. | Reddy, P. G.; Varghese, B.; Baskaran, S. Org. Lett. 2003, 5, 583–585. doi:10.1021/ol027563v |
| 14. | Carbonnel, S.; Troin, Y. Heterocycles 2002, 57, 1807–1830. |
| 15. | Zaminer, J.; Stapper, C.; Blechert, S. Tetrahedron Lett. 2002, 43, 6739–6741. doi:10.1016/S0040-4039(02)01534-4 |
| 16. | Corvo, M.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X |
| 50. | Bardot, V.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C. Tetrahedron: Asymmetry 1997, 8, 1111–1114. doi:10.1016/S0957-4166(97)00079-7 |
| 8. | Michael, J. P.; Gravestock, D. Eur. J. Org. Chem. 1998, 865–870. doi:10.1002/(SICI)1099-0690(199805)1998:5<865::AID-EJOC865>3.0.CO;2-3 |
| 19. | Chalard, P.; Remuson, R.; Gelas-Mialhe, Y.; Gramain, J. C.; Canet, I. Tetrahedron Lett. 1999, 40, 1661–1664. doi:10.1016/S0040-4039(99)00059-3 |
| 51. | Gardette, D.; Gelas-Mialhe, Y.; Gramain, J. C.; Perrin, B.; Remuson, R. Tetrahedron: Asymmetry 1998, 9, 1823–1838. doi:10.1016/S0957-4166(98)00170-0 |
| 32. | Malching, K. H.; Hiemstra, H.; Klaver, W. J.; Speckamp, W. N. Tetrahedron Lett. 1986, 27, 4799–4802. doi:10.1016/S0040-4039(00)85068-6 |
| 33. | Klaver, W. J.; Hiemstra, H.; Speckamp, W. N. Tetrahedron Lett. 1987, 28, 1581–1584. doi:10.1016/S0040-4039(01)81047-9 |
| 69. | Bardot, V. Ph.D. Thesis, Université Clermont-Ferrand, Clermont-Ferrand, France, 1994. |
| 34. | Conchon, E.; Gelas-Mialhe, Y.; Remuson, R. Tetrahedron: Asymmetry 2006, 17, 1253–1257. doi:10.1016/j.tetasy.2006.04.027 |
| 17. | Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X |
| 5. | Slosse, P.; Hootelé, C. Tetrahedron 1981, 37, 4287–4294. doi:10.1016/0040-4020(81)85024-7 |
| 42. | King, F. D. J. Chem. Soc., Perkin Trans. 1 1986, 447–453. doi:10.1039/p19860000447 |
| 43. | Comins, D. L.; LaMunyon, D. H. J. Org. Chem. 1992, 57, 5807–5809. doi:10.1021/jo00048a003 |
| 44. | Pilli, R. A.; Dias, L. C.; Maldaner, A. O. J. Org. Chem. 1995, 60, 717–722. doi:10.1021/jo00108a040 |
| 43. | Comins, D. L.; LaMunyon, D. H. J. Org. Chem. 1992, 57, 5807–5809. doi:10.1021/jo00048a003 |
| 45. | Davis, F. A.; Xu, H.; Zhang, J. J. Org. Chem. 2007, 72, 2046–2052. doi:10.1021/jo062365t |
| 17. | Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1991, 2, 183–186. doi:10.1016/S0957-4166(00)82354-X |
| 4. | Slosse, P.; Hootelé, C. Tetrahedron Lett. 1978, 397–398. doi:10.1016/S0040-4039(01)85135-2 |
| 5. | Slosse, P.; Hootelé, C. Tetrahedron 1981, 37, 4287–4294. doi:10.1016/0040-4020(81)85024-7 |
| 39. | De Saboulin Bollena, A.; Gelas-Mialhe, Y.; Gramain, J. C.; Perret, A.; Remuson, R. J. Nat. Prod. 2004, 67, 1029–1031. doi:10.1021/np049902u |
| 40. | Kobayashi, T.; Hasegawa, F.; Katsunori, T.; Katsumura, S. Org. Lett. 2006, 8, 5917–5921. doi:10.1021/ol0622716 |
| 41. | Kobayashi, T.; Hasegawa, F.; Tanaka, K.; Katsumura, S. Org. Lett. 2006, 8, 3813–3816. doi:10.1021/ol0614065 |
| 36. | Lüning, B.; Lundin, C. Acta Chem. Scand. 1967, 21, 2136–2142. |
| 37. | Sonnet, P. E.; Netzel, D. A.; Mendoza, R. J. Heterocycl. Chem. 1979, 16, 1041–1047. |
| 38. | Blomquist, L.; Leander, K.; Lüning, B.; Rosemblom, J. Acta Chem. Scand. 1972, 26, 3203–3206. |
© 2007 Remuson; licensee Beilstein-Insitut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)