Abstract
Two-directional synthesis represents an ideal strategy for the rapid elaboration of simple starting materials and their subsequent transformation into complex molecular architectures. As such, it is becoming recognised as an enabling technology for diversity-oriented synthesis. Herein, we provide a thorough account of our work combining two-directional synthesis with diversity-oriented synthesis, with particular reference to the synthesis of polycyclic alkaloid scaffolds.

Graphical Abstract
Introduction
Diversity-oriented synthesis (DOS) aims to prepare structurally diverse compound collections in an efficient manner [1-3]. Of the possible “types of diversity” that can be incorporated into a compound collection, the most important, in terms of creating a functionally (biologically) diverse collection, is generally considered to be scaffold (or skeletal) diversity, i.e., the variation of molecular frameworks between compounds [4,5]. Therefore, one of the key challenges in DOS is the development of strategies that allow the efficient generation of a range of complex molecular scaffolds. A large number of approaches towards this goal have been reported, with some of the most effective being based around the “folding-up” of functionalised linear substrates into cyclic molecular architectures [6-8]. The design and synthesis of these linear substrates can, in itself, represent a significant challenge as it is desirable that these compounds are easily accessible in a small number of synthetic steps. Two-directional synthesis [9-12] offers a powerful method for the synthesis of such substrates, because each synthetic transformation has the potential to provide twice as much molecular complexity compared to standard approaches.
We have recently reported a strategy for DOS that combined two-directional synthesis with the use of these folding reaction pathways [13]. In this work, two-directional synthesis was used to rapidly generate a series of linear aminoalkenes, which were then folded into bicyclic and tricyclic scaffolds through Lewis acid mediated cascade processes. The compounds produced in this campaign were reminiscent of naturally occurring alkaloids, such as the Coccinellidae natural products, which are secreted by ladybirds to deter predators [14]. The total synthesis of one of their number, myrrhine, was also achieved by the elaboration of one of the compounds produced. In this article, the work is presented in more detail, alongside additional results from our work combining two-directional synthesis with DOS. Treated together, we believe these works provide useful insights into the potential utility of two-directional synthesis as an enabling technology for DOS.
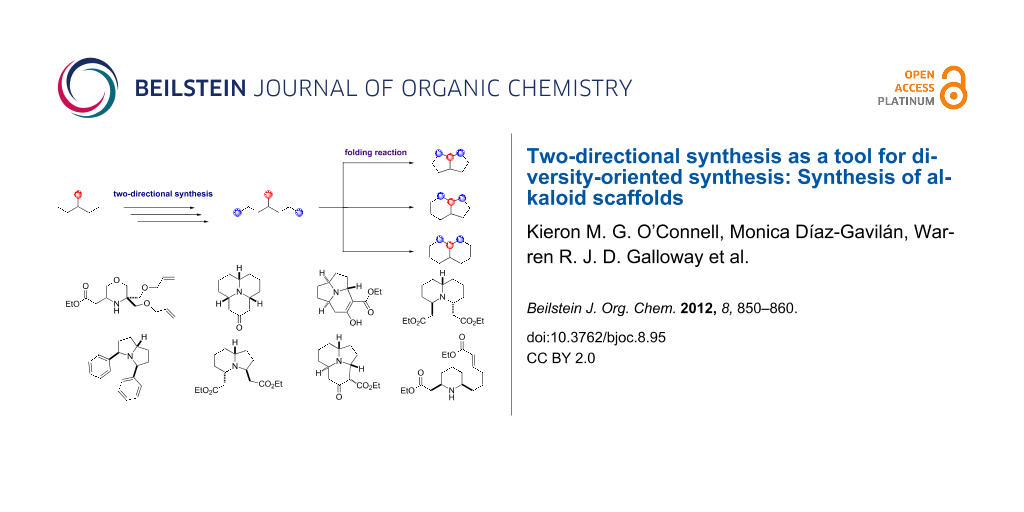
The initial DOS campaign was largely inspired by the pioneering work of Robert Stockman on combining two-directional synthesis with tandem reactions to create complex molecular architectures [15-18]. The key folding step in the DOS was the Lewis acid mediated pairing reaction of a nucleophilic amino group with suitable electrophilic functionality, provided by Michael acceptor α,β-unsaturated ester groups. Two-directional synthesis was used to append these electrophilic groups at two positions around the linear substrates, allowing bicyclisation processes to be instigated. The scaffold diversity between the products then resulted from the different ring sizes that it was possible to form from these linear substrates. (Figure 1 shows an overview of the DOS strategy.)
![[1860-5397-8-95-1]](/bjoc/content/figures/1860-5397-8-95-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Synthesis of linear precursors
Our initial studies explored the synthesis and reactivity of four N-Boc-aminoalkenes containing α,β-unsaturated ester groups (1–4, Scheme 1), as substrates for intramolecular pairing reactions. The N-Boc protecting group was chosen to give the potential for deprotection to be carried out in tandem with the Lewis acid catalysed cyclisation reactions by intramolecular conjugate addition. Three of these compounds (1–3) were obtained from the corresponding alcohols [19,20] in three steps: Mitsunobu reaction with NH-Boc-tosylate, followed by tosyl deprotection with magnesium, and finally two-directional cross metathesis with ethyl acrylate to install the desired α,β-unsaturated ester functionality. This sequence provided the desired N-Boc-aminoalkenes in respectable overall yields of 38–56%. Compound 4 was prepared in a four-step sequence from the requisite phenyldialkyl alcohol. Ritter reaction with chloroacetonitrile followed by cleavage of the resulting chloroacetamide with thiourea gave the free amine [21], which was then protected with Boc anhydride. Finally, cross metathesis with ethyl acrylate furnished the desired compound 4 in 24% overall yield.
![[1860-5397-8-95-i1]](/bjoc/content/inline/1860-5397-8-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of linear cyclisation precursors 1–4.
Scheme 1: Synthesis of linear cyclisation precursors 1–4.
Cyclisation reactions
The first attempts at the tandem Boc-deprotection/bicyclisation of these substrates were performed by using AlCl3 as the Lewis acid (Scheme 2); compounds 1–4 were treated with 1.1 equiv of AlCl3 in dichloromethane at room temperature. These conditions proved effective at promoting bicyclisation for compounds 1, 2 and 4, for which the desired bicyclic products were obtained in 67–85% yield, as a mixture of diastereomers. The cyclisation of 1 gave pyrrolizidine 5 as a mixture of 4,10-trans-7,10-trans (trans-5) (42%) and 4,10-cis-7,10-trans (cis-5) (28%) isomers, which proved to be separable by flash chromatography. The stereochemistry of cis-5 was confirmed by analogy to known 1H and 13C NMR values [22] and by NOESY spectroscopy, which showed enhancements between H-7, H-4 and H-10. In this case, it proved possible to achieve an improved yield of both diastereomers by treating 1 with an excess (163 equiv) of trifluroacetic acid, which furnished trans-5 and cis-5 in 53% and 34% yield, respectively. The corresponding reaction of phenyl-substituted analogue 4 also gave a mixture of two diastereomers; the major diastereomer was 4,10-trans-7,10-cis isomer (trans-6), which was formed in 50% yield, and a 17% yield of the 4,7,10-cis-isomer (cis-6) was also obtained.
![[1860-5397-8-95-i2]](/bjoc/content/inline/1860-5397-8-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: AlCl3 catalysed tandem Boc-removal/bicyclisation processes; the yields quoted refer to the isolated yields of single compounds.
Scheme 2: AlCl3 catalysed tandem Boc-removal/bicyclisation processes; the yields quoted refer to the isolated...
Indolizidine 7 was produced in very good overall yield (85%). The unsymmetrical nature of this compound gave rise to the possibility of formation of additional diastereomers when compared to 5 and 6; however, once again only two were formed in any appreciable amount. The compounds obtained were both found to have a trans-fused geometry at the ring junction, as indicated by IR (strong absorbance at 2850 cm−1) and 1H NMR (H-7 chemical shift around 2.4 ppm) spectroscopy [23,24]. The ester-bearing side chains were also found to be trans to each other in both cases, meaning that the two products obtained differ from each other only in which ring has the side chain cis to the ring junction hydrogen. The favoured product was the 4,11-trans-7,11-cis isomer (trans-7) in which the side chain of the 6-membered ring is cis to the ring junction proton; this compound was isolated in 55% yield. The alternative 4,11-trans-4,7-cis isomer (trans’-7) was obtained in 30% yield. Despite the good yields obtained for these three examples, the cyclisation of 3 under these conditions proved disappointing, with 4,12-trans-8,12-cis-quinolizidine (trans-8) only obtained in 18% yield, along with 40% of monocyclic species 9.
In light of the difficulties encountered in producing the desired bicyclic species, an optimisation study of the cyclisation of 3 was undertaken, which resulted in a number of interesting findings that are summarised in Table 1. The initial alterations made little difference to the process; increasing the reaction time up to seven days had essentially no effect on the product ratio, with trans-8 and 9 obtained in 21% and 37% yield, respectively, and increasing the temperature to the point of reflux in dichloromethane also had little effect on the conversion. However, when the reaction solvent was changed to toluene and the reaction mixture heated to reflux, trans-8 and 9 were still formed in similar proportions (24% and 21%), but in this case an additional product was also isolated in 30% yield. This compound was found to be tricyclic compound 10 (Scheme 3a), which was obtained as a single diastereomer with all of the ring junction protons on the same face. This all–cis-stereochemistry was surprising, as so far 8 had only been obtained with the side chains in trans- configuration; however the configuration of 10 was unambiguously confirmed by X-ray crystallography. In some ways the formation of a tricyclic species such as 10 was not altogether surprising, as a similar tricyclic species was generated in Stockman’s synthesis of hippodamine [25]. In that work, bicycle trans-8 was transformed into the corresponding tricyclic compound (possessing cis–trans ring junction stereochemistry) by a base-mediated Dieckmann cyclisation (Scheme 3b). It seems in our case that, under the correct conditions, a Dieckmann reaction can be made to occur in tandem with the Boc-deprotection and double-conjugate addition processes. This one-pot, four-step reaction process is extremely interesting for the amount of molecular complexity generated in a single transformation, and also because the all-cis-stereochemistry of 10, which differs from the cis–trans-stereochemistry observed by Stockman for the Dieckman cyclisation of trans-8. For these reasons, further investigations into the process were carried out.
Table 1: Overview of the Lewis acid mediated folding reactions of 3.
| solvent |
Lewis acid
(equiv) |
temp. | % yield | |||
|---|---|---|---|---|---|---|
| trans-8 | cis-8 | 9 | 10 | |||
| DCMa | AlCl3 (1.1) | rt | 18 | — | 40 | — |
| DCMb | AlCl3 (1.1) | rt | 21 | — | 37 | — |
| toluenea | AlCl3 (1.1) | reflux | 24 | — | 21 | 30 |
| toluenea | AlCl3 (3) | reflux | 43 | — | 13 | — |
| toluenea | Sc(OTf)3 (3) | reflux | 50 | — | 21 | — |
| toluenea | Sn(OTf)2 (0.5) | reflux | — | 9 | — | 72 |
| toluenea | Sn(OTf)2 (1.1) | reflux | 30 | 13 | — | 23 |
| toluene | Sn(OTf)2 (3) | reflux | 29 | — | 10 | — |
aReaction stirred overnight; breaction stirred for seven days.
![[1860-5397-8-95-i3]](/bjoc/content/inline/1860-5397-8-95-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: (a) AlCl3 catalysed formation of tricyclic alkaloid 10 along with an X-ray crystal structure of 10; (b) base-mediated Dieckmann cyclisation of trans-8 employed by Stockman and co-workers in the synthesis of hippodamine [25].
Scheme 3: (a) AlCl3 catalysed formation of tricyclic alkaloid 10 along with an X-ray crystal structure of 10;...
The amount of AlCl3 was increased to 3 equiv; however, this led to the suppression of tricycle formation in favour of a slight increase in the yield of trans-8 to 43% (a 13% yield of 9 was also obtained). Switching the Lewis acid to Sc(OTf)3 and still using 3 equiv, gave a slight improvement in the yields of the trans-8 to 50% and 9 to 21% but no formation of 10. Switching the Lewis acid again to Sn(OTf)2 resulted in a decrease in the yields of trans-8 to 29% and 9 to 10% and again no formation of 10. The amount of Lewis acid was then reduced to 0.5 equiv, which dramatically altered the course of the reaction. Performing the reaction under reflux in toluene with 0.5 equiv Sn(OTf)2 produced tricycle 10 in 72% isolated yield. Thus both trans-8 and 10 could be accessed in good yields from the same substrate simply by varying the amount and identity of the Lewis acid used.
Interestingly, the catalytic variant of the reaction also produced 9% of the bicyclic cis-8, which had not been isolated from any of the previous reactions. For completeness, one further reaction was performed by using 1.1 equiv of Sn(OTf)2; careful purification of this reaction gave 23% of 10, 30% of trans-8 and 13% of cis-8. The presence of the previously undetected cis-8 in these two final reactions was intriguing and led us to speculate as to whether a different mechanistic pathway could be in operation depending on the amount of Lewis acid used.
A number of factors led to this mechanistic speculation; principal among them was the fact that cis-8 was never detected in reactions in which 10 was not formed. In all of the earlier experiments the only bicycle detected was trans-8, implying that the double-conjugate addition process heavily favours the formation of this compound. Therefore it was considered that cis-8 could be forming from 10; suggesting, somewhat counter intuitively, that the Dieckmann cyclisation to give 6,10-bridged bicycle 11 could in some cases be favoured over the expected double-conjugate addition. Transannular conjugate addition across the 10-membered ring of 11 would give 10, and a retro-Dieckmann process could then form cis-8. Several control experiments were run in an attempt to validate this hypothesis (Scheme 4a).
![[1860-5397-8-95-i4]](/bjoc/content/inline/1860-5397-8-95-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) Optimal conditions to obtain trans-8 and 10 and the control experiments carried out to probe the mechanism of the process; (b) proposed mechanistic pathway for the tricyclisation of 3.
Scheme 4: (a) Optimal conditions to obtain trans-8 and 10 and the control experiments carried out to probe th...
Trans-8 was treated with both stoichiometric and catalytic amounts of Sn(OTf)2 and there was no evidence of tricycle formation in either case, with only starting material recovered from the reactions. The direct formation of 10 from trans-8 was not thought to be possible due to their differing stereochemistry; however, an alternative diastereomer of 10 may be expected to form (as observed by Stockman for the corresponding base-mediated process) [25]. Monocycle 9 was treated with catalytic Sn(OTf)2, which led to a mixture of trans-8 and 10. As the formation of 10 from trans-8 had been proven not to occur, it seemed reasonable to assume from this that it is possible to form both species from 9. It is also noteworthy that the best yields of 10 were obtained when the reaction was performed while fitted with a Dean–Stark apparatus containing pieces of sodium to trap the ethanol formed in the Dieckmann cyclisation, and thus inhibit the retro-Dieckmann reaction. Finally, 10 was treated with catalytic Sn(OTf)2, which, as expected, gave cis-8, suggesting that the retro-Dieckmann reaction does occur.
Taking these results into account, we tentatively suggest that the mechanism for the tricyclisation of 3 does indeed proceed via a Dieckmann cyclisation to form the 10-membered ring followed by transannular conjugate addition (Scheme 4b). The fact that this appears to occur when lower amounts of Lewis acid are used (0.5 or 1.1 equiv) but not when three equiv are used indicates that the mechanistic path of the cascade is dependent on the amount of the Lewis acid present. When three equiv of Lewis acid are present (the “stoichiometric” process), it is feasible that during the course of the reaction both of the carbonyl groups are activated simultaneously, allowing the double-conjugate addition to proceed smoothly. However, when fewer equiv are used (the “catalytic” process) there is a relative deficiency of Lewis acid present, and thus, it is less probable that both carbonyl groups can be coordinated to separate Lewis acid molecules simultaneously. If the Dieckmann reaction occurs via a standard 6-membered transition state, it requires both carbonyls to be either coordinated to, or bonded to, a single metal centre, and so this could go some way towards explaining the apparent course of the reaction. Assuming that the Lewis acid remains bonded to the carbonyl of the enol form of the ester group after the first conjugate addition, there may be insufficient Lewis acid free in solution to activate the second ester group separately, leaving it effectively inert to conjugate addition (and Dieckmann cyclisation). However, if the second carbonyl group becomes coordinated to the metal centre that is bonded to the first, the Dieckmann reaction becomes the most favourable process and so occurs preferentially to the second conjugate addition. For these reasons, we cautiously postulate that the Dieckmann reaction may occur via a chelated transition state such as 12, in which both carbonyls are coordinated to a single metal centre.
The data in Table 1 provides further support for the suggestion that different mechanisms can operate depending on the amount of Lewis acid used. This support is provided by the fact that the formation of 10 proceeds best under truly catalytic conditions: when 0.5 equiv Sn(OTf)2 were used, a 72% yield of 10 was obtained compared to 23% when 1.1 equiv were used. In fact, it appears that when 1.1 equiv of Lewis acid are used both mechanisms can occur, as indicated by the formation of both trans-8 and 10 in these reactions, but when 0.5 equiv are used the formation of trans-8 is not observed suggesting that under these conditions the simple double-conjugate addition cannot occur. The identity of the Lewis acid used in the reaction seems not to overtly affect the course of the reaction in terms of the products obtained, as similar product distributions were observed for the reactions using three equiv of AlCl3, Sc(OTf)3 and Sn(OTf)2. The formation of 10 was also not limited to the use of Sn(OTf)2, as a 30% yield of 10 was obtained for the reaction using 1.1 equiv of AlCl3.
Between them, the catalytic and stoichiometric variants of this reaction provide a useful illustration of the use of reagent-based diversification within a predominantly substrate-based strategy. Attempts were then made to apply this reagent-based diversification to the other linear substrates (Scheme 5). Compound 2 was treated with 0.5 equiv of Sn(OTf)2 in acetonitrile, which surprisingly did not lead to the formation of the 6-6-5-tricyclic species, instead producing trans’-7 in 69% yield along with 10% trans-7. While this reaction did not produce any tricyclic species, it was an interesting result, as the selectivity for trans’-7 over trans-7 was the opposite of that observed for the original AlCl3 catalysed process. Performing the reaction in toluene with 1.1 equiv of Sn(OTf)2 did produce the expected tricyclic species in 50% yield as a mixture of two diastereomers (cis-13 and trans-13) in a 4:1 ratio, proving that the folding of this linear substrate into tricyclic species in one-pot was also possible. Unfortunately, this did not prove to be the case for 1; all attempts to transform 1 into a tricyclic species in one pot, using either stoichiometric or catalytic Lewis acid were unsuccessful. However, it did prove possible to perform the Dieckmann reaction on the bicyclic compounds cis-5 and trans-5 to access the tricyclic architectures. The bicyclic species were treated with LDA at −18 °C, which affected the desired cyclisation in both cases. Strangely, the process proved far more efficient for trans-5, giving trans-14 in 91% yield, compared to 38% for the cis-isomer. In both cases the 1H NMR suggested that these compounds exist as the usually disfavoured enol tautomer.
![[1860-5397-8-95-i5]](/bjoc/content/inline/1860-5397-8-95-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: DOS of 5-5-6 and 5-6-6 tricyclic alkaloids 13 and 14.
Scheme 5: DOS of 5-5-6 and 5-6-6 tricyclic alkaloids 13 and 14.
These reactions, along with the reactions of 1, clearly illustrate the power of this two-directional approach to DOS. Using this methodology it was possible to access five bicyclic and tricyclic scaffolds covering a range of 3D shapes, including pyrrolizidines, indolizidines and quinolizidines, along with 6-6-6 and 5-6-6 azatricyclic species in a single transformation from a small collection of structurally simple linear starting materials. One further tricyclic scaffold (5-5-6) was also accessible in one further synthetic step. The effective introduction of reagent-based diversification into the strategy was extremely satisfying, as by altering the choice of Lewis acid and reaction temperature we were able to adjust the course and selectivity of the reactions to generate different scaffolds and stereochemistry. This combination of reagent and substrate-based approaches for the generation of molecular diversity can afford many interesting possibilities not achievable by either approach alone.
Total synthesis of myrrhine
Inspired by Stockman’s syntheses of the related alkaloids hippodamine and epi-hippodamine [26], tricyclic species 10 was identified as a potential intermediate for the total synthesis of myrrhine, which was then achieved in three steps from 10 (Scheme 6). Compound 10 was treated with Na2CO3 in a mixture of EtOH and H2O under reflux to achieve ester saponification, which was followed by decarboxylation, proceeding smoothly to give the corresponding ketone in 76% yield. The ketone was then transformed to the exocyclic alkene 15 in 61% yield by Wittig reaction with the appropriate phosphonium salt. The final step in the synthesis was a diastereoselective reduction of the double bond with hydrogen gas and Raney-nickel. This was achieved in a moderate 57% yield and with good (~10:1) diastereoselectivity by complexing the nitrogen lone pair with tosic acid, effectively blocking the undesirable face of the tricycle during the course of the reduction. This reduction also produced 5% of the unnatural (or as yet undiscovered) isomer epi-myrrhine. The N-oxide of myrrhine, which has also not been isolated from natural sources, was then synthesised in 96% yield by treating myrrhine with mCPBA. This synthesis of myrrhine compares favourably with previously reported syntheses [26-28], achieving the feat in eight steps and 7% overall yield.
![[1860-5397-8-95-i6]](/bjoc/content/inline/1860-5397-8-95-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Total synthesis of myrrhine, epi-myrrhine and myrrhine-N-oxide.
Scheme 6: Total synthesis of myrrhine, epi-myrrhine and myrrhine-N-oxide.
Alternative starting materials
The evident efficiency of two-directional synthesis in a DOS context, as exemplified by our synthesis of these alkaloid scaffolds, has led us to continue investigations in this area and to explore the potential utility of this approach for a range of different substrates. Among these substrates, two that stand out as particularly promising are nitromethane and tris(hydroxymethyl)aminomethane (Tris) 16.
Nitromethane is of great interest to us as a potential DOS substrate, as we have a long-standing interest in developing divergent reaction pathways from small and simple starting materials [29,30]. As it represents a one-carbon unit, nitromethane is an ideal substrate for investigation. For Tris, the central quarternary carbon centre is the key point of interest, as we believe that, with the judicious selection of appendages, it should allow us to access a range of bicyclic structures, including examples of fused, bridged and spiro bicycles. Preliminary studies into the utility of these substrates in a DOS context have yielded some promising results.
Our work with nitromethane has so far led to the synthesis of meso-diphenylpyrrolizidine 17, which was achieved in three steps (Scheme 7). Nitromethane was treated with NaOH, and the resulting anion was used to displace the chloride from 3-chloro-1-phenylpropan-1-one giving a 90% yield of the nitroketone. Two-directional synthesis of diketone 18 in this fashion did not prove to be feasible; however, it was achieved in good yield by Michael addition of the nitroketone anion to phenylvinylketone. Subjecting diketone 18 to H2 gas and Raney-nickel then reduced the nitro group and effected the desired double reductive amination to give 17 in 30% yield. It is likely that the scope of this sequence could be extended to include indolizidine and quinolizidine scaffolds, and so provide an alternative route to these frameworks, instead of the double Michael addition strategy.
![[1860-5397-8-95-i7]](/bjoc/content/inline/1860-5397-8-95-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Use of nitromethane in DOS: Synthesis of meso-diphenylpyrrolidizine 17.
Scheme 7: Use of nitromethane in DOS: Synthesis of meso-diphenylpyrrolidizine 17.
The three hydroxy and single amino groups of Tris give the potential for many variations in substitution; however, our studies have so far focused on allylated derivatives, in particular the triallyl derivative 19 (Scheme 8). The synthesis of 19 was achieved in two steps from Tris: N-Boc protection proceeded in 70% yield, and was followed by alkylation with an excess of allyl bromide to provide the desired triallyl species in 66% yield. Cross metathesis of 19 with ethyl acrylate was then performed. Fortunately, it proved to be possible to achieve some selectivity for different products by varying the catalyst used. Treating 19 with 3% Grubbs II catalyst in neat ethyl acrylate at room temperature gave monoester 20 in 41% yield, whereas performing the reaction with 5% Hoveyda-Grubbs II catalyst gave a 73% yield of triester 21.
![[1860-5397-8-95-i8]](/bjoc/content/inline/1860-5397-8-95-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Use of Tris as a substrate for DOS: Synthesis of decorated morpholines 22 and 23.
Scheme 8: Use of Tris as a substrate for DOS: Synthesis of decorated morpholines 22 and 23.
These two metathesis products were then subjected to the tandem deprotection-cyclisation conditions, this time by using two equiv of AlCl3 in dichloromethane at room temperature. For monoester 20, only a single conjugate addition was possible, leaving the pendant terminal alkene groups as handles for further reactivity. This cyclisation proceeded to give the expected, functionalised morpholine scaffold 22 in a moderate 33% yield. A similar product was also obtained for 21, with a single conjugate addition process occurring to give 23 in 73% yield and no trace of the bicyclic product detected. A number of attempts were then made to force the reaction to occur, by varying the Lewis acid to Sn(OTf)2 and Sc(OTf)3 and by heating under reflux in toluene, all of which produced varying amounts of 23, with no trace of the bicyclic product detected. This result was disappointing and also, to some degree, surprising given the relative ease of formation of the corresponding quinolizidine compounds. We speculate that the relative difficulty in the reaction is due to the reduced availability of the lone pair of the morpholine nitrogen compared to the piperidine nitrogen (pKa 8.36 versus 11.22) [31], retarding the rate of nucleophilic attack. Another possibility is that the oxygens in the pendant chains can form a stable hydrogen-bonded species with the NH group that inhibits the reaction of the nitrogen with the Michael acceptor ester groups. Studies involving the use of Tris as a potential DOS substrate remain on-going within our laboratories.
Conclusion
The work presented in this article serves to illustrate the potential power of two-directional synthesis in DOS. The use of two-directional synthesis allowed us to access a range of bicyclic and tricyclic molecular scaffolds, rapidly and efficiently, by following a common reaction scheme. The nature of the two-directional synthesis lends itself to the formation of bicyclic compounds by the folding up of doubly substituted precursors, and it proved to be a very effective strategy for the synthesis of natural-product-like alkaloid scaffolds. Our work so far in this area has focused mainly on the synthesis of fused bicyclic compounds; however, we hope in the future to be able to apply a two-directional synthesis approach to the DOS of a wide range of molecular scaffolds and structural classes.
References
-
Galloway, W. R. J. D.; Isidro-Llobet, A.; Spring, D. R. Nat. Commun. 2010, 1, No. 80. doi:10.1038/ncomms1081
Return to citation in text: [1] -
Nielsen, T. E.; Schreiber, S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. doi:10.1002/anie.200703073
Return to citation in text: [1] -
Spandl, R. J.; Díaz-Gavilán, M.; O’Connell, K. M. G.; Thomas, G. L.; Spring, D. R. Chem. Rec. 2008, 8, 129–142. doi:10.1002/tcr.20144
Return to citation in text: [1] -
Galloway, W. R. J. D.; Díaz-Gavilán, M.; Isidro-Llobet, A.; Spring, D. R. Angew. Chem., Int. Ed. 2009, 48, 1194–1196. doi:10.1002/anie.200805452
Return to citation in text: [1] -
Dow, M.; Fisher, M.; James, T.; Marchetti, F.; Nelson, A. Org. Biomol. Chem. 2012, 10, 17–28. doi:10.1039/c1ob06098h
Return to citation in text: [1] -
Burke, M. D.; Berger, E. M.; Schreiber, S. L. Science 2003, 302, 613–618. doi:10.1126/science.1089946
Return to citation in text: [1] -
Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486
Return to citation in text: [1] -
O’Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563–9571. doi:10.1002/chem.201000707
Return to citation in text: [1] -
Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002
Return to citation in text: [1] -
Vrettou, M.; Gray, A. A.; Brewer, A. R. E.; Barrett, A. G. M. Tetrahedron 2007, 63, 1487–1536. doi:10.1016/j.tet.2006.09.109
Return to citation in text: [1] -
Kennedy, A.; Nelson, A.; Perry, A. Beilstein J. Org. Chem. 2005, 1, No. 2. doi:10.1186/1860-5397-1-2
Return to citation in text: [1] -
Gignoux, C.; Newton, A. F.; Barthelme, A.; Lewis, W.; Alcaraz, M.-L.; Stockman, R. A. Org. Biomol. Chem. 2012, 10, 67–69. doi:10.1039/c1ob06380d
Return to citation in text: [1] -
Díaz-Gavilán, M.; Galloway, W. R. J. D.; O'Connell, K. M. G.; Hodkingson, J. T.; Spring, D. R. Chem. Commun. 2010, 46, 776–778. doi:10.1039/b917965h
Return to citation in text: [1] -
Happ, G. M.; Eisner, T. Science 1961, 134, 329–331. doi:10.1126/science.134.3475.329
Return to citation in text: [1] -
Stockman, R. A. Tetrahedron Lett. 2000, 41, 9163–9165. doi:10.1016/S0040-4039(00)01640-3
Return to citation in text: [1] -
Stockman, R. A.; Sinclair, A.; Arini, L. G.; Szeto, P.; Hughes, D. L. J. Org. Chem. 2004, 69, 1598–1602. doi:10.1021/jo035639a
Return to citation in text: [1] -
Rejzek, M.; Stockman, R. A.; van Maarseveen, J. H.; Hughes, D. L. Chem. Commun. 2005, 4661–4662. doi:10.1039/b508969g
Return to citation in text: [1] -
Robbins, D.; Newton, A. F.; Gignoux, C.; Legeay, J.-C.; Sinclair, A.; Rejzek, M.; Laxon, C. A.; Yalamanchili, S. K.; Lewis, W.; O’Connell, M. A.; Stockman, R. A. Chem. Sci. 2011, 2, 2232–2235. doi:10.1039/c1sc00371b
Return to citation in text: [1] -
Boyer, F.-D.; Hanna, I. Eur. J. Org. Chem. 2006, 471–482. doi:10.1002/ejoc.200500645
Return to citation in text: [1] -
Schwartz, B. D.; McErlean, C. S. P.; Fletcher, M. T.; Mazomenos, B. E.; Konstantopoulou, M. A.; Kitching, W.; De Voss, J. J. Org. Lett. 2005, 7, 1173–1176. doi:10.1021/ol050143w
Return to citation in text: [1] -
Jirgensons, A.; Kauss, V.; Kalvinsh, I.; Gold, M. R. Synthesis 2000, 1709–1712. doi:10.1055/s-2000-8208
Return to citation in text: [1] -
Scarpi, D.; Occhiato, E. G.; Guarna, A. J. Org. Chem. 1999, 64, 1727–1732. doi:10.1021/jo981946i
Return to citation in text: [1] -
Crabb, T. A.; Newton, R. F.; Jackson, D. Chem. Rev. 1971, 71, 109–126. doi:10.1021/cr60269a005
Return to citation in text: [1] -
Rader, C. P.; Young, R. L., Jr.; Aaron, H. S. J. Org. Chem. 1965, 30, 1536–1539. doi:10.1021/jo01016a048
Return to citation in text: [1] -
Rejzek, M.; Stockman, R. A.; Hughes, D. L. Org. Biomol. Chem. 2005, 3, 73–83. doi:10.1039/b413052a
Return to citation in text: [1] [2] [3] -
Ayer, W. A.; Dawe, R.; Eisner, R. A.; Furuichi, K. Can. J. Chem. 1976, 54, 473–481. doi:10.1139/v76-064
Return to citation in text: [1] [2] -
Mueller, R. H.; Thompson, M. E.; DiPardo, R. M. J. Org. Chem. 1984, 49, 2217–2231. doi:10.1021/jo00186a029
Return to citation in text: [1] -
Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476–2484. doi:10.1021/jo062533h
Return to citation in text: [1] -
Wyatt, E. E.; Fergus, S.; Galloway, W. R. J. D.; Bender, A.; Fox, D. J.; Plowright, A. T.; Jessiman, A. S.; Spring, D. R. Chem. Commun. 2006, 3296–3298. doi:10.1039/b607710b
Return to citation in text: [1] -
Thomas, G. L.; Spandl, R. J.; Glansdorp, F. G.; Welch, M.; Bender, A.; Cockfield, J.; Lindsay, J. A.; Bryant, C.; Brown, D. F. J.; Loiseleur, O.; Rudyk, H.; Ladlow, M.; Spring, D. R. Angew. Chem., Int. Ed. 2008, 47, 2808–2812. doi:10.1002/anie.200705415
Return to citation in text: [1] -
Hall, H. K., Jr. J. Am. Chem. Soc. 1957, 79, 5441–5444. doi:10.1021/ja01577a030
Return to citation in text: [1]
| 29. | Wyatt, E. E.; Fergus, S.; Galloway, W. R. J. D.; Bender, A.; Fox, D. J.; Plowright, A. T.; Jessiman, A. S.; Spring, D. R. Chem. Commun. 2006, 3296–3298. doi:10.1039/b607710b |
| 30. | Thomas, G. L.; Spandl, R. J.; Glansdorp, F. G.; Welch, M.; Bender, A.; Cockfield, J.; Lindsay, J. A.; Bryant, C.; Brown, D. F. J.; Loiseleur, O.; Rudyk, H.; Ladlow, M.; Spring, D. R. Angew. Chem., Int. Ed. 2008, 47, 2808–2812. doi:10.1002/anie.200705415 |
| 31. | Hall, H. K., Jr. J. Am. Chem. Soc. 1957, 79, 5441–5444. doi:10.1021/ja01577a030 |
| 1. | Galloway, W. R. J. D.; Isidro-Llobet, A.; Spring, D. R. Nat. Commun. 2010, 1, No. 80. doi:10.1038/ncomms1081 |
| 2. | Nielsen, T. E.; Schreiber, S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. doi:10.1002/anie.200703073 |
| 3. | Spandl, R. J.; Díaz-Gavilán, M.; O’Connell, K. M. G.; Thomas, G. L.; Spring, D. R. Chem. Rec. 2008, 8, 129–142. doi:10.1002/tcr.20144 |
| 13. | Díaz-Gavilán, M.; Galloway, W. R. J. D.; O'Connell, K. M. G.; Hodkingson, J. T.; Spring, D. R. Chem. Commun. 2010, 46, 776–778. doi:10.1039/b917965h |
| 26. | Ayer, W. A.; Dawe, R.; Eisner, R. A.; Furuichi, K. Can. J. Chem. 1976, 54, 473–481. doi:10.1139/v76-064 |
| 9. | Poss, C. S.; Schreiber, S. L. Acc. Chem. Res. 1994, 27, 9–17. doi:10.1021/ar00037a002 |
| 10. | Vrettou, M.; Gray, A. A.; Brewer, A. R. E.; Barrett, A. G. M. Tetrahedron 2007, 63, 1487–1536. doi:10.1016/j.tet.2006.09.109 |
| 11. | Kennedy, A.; Nelson, A.; Perry, A. Beilstein J. Org. Chem. 2005, 1, No. 2. doi:10.1186/1860-5397-1-2 |
| 12. | Gignoux, C.; Newton, A. F.; Barthelme, A.; Lewis, W.; Alcaraz, M.-L.; Stockman, R. A. Org. Biomol. Chem. 2012, 10, 67–69. doi:10.1039/c1ob06380d |
| 26. | Ayer, W. A.; Dawe, R.; Eisner, R. A.; Furuichi, K. Can. J. Chem. 1976, 54, 473–481. doi:10.1139/v76-064 |
| 27. | Mueller, R. H.; Thompson, M. E.; DiPardo, R. M. J. Org. Chem. 1984, 49, 2217–2231. doi:10.1021/jo00186a029 |
| 28. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476–2484. doi:10.1021/jo062533h |
| 6. | Burke, M. D.; Berger, E. M.; Schreiber, S. L. Science 2003, 302, 613–618. doi:10.1126/science.1089946 |
| 7. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 8. | O’Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563–9571. doi:10.1002/chem.201000707 |
| 25. | Rejzek, M.; Stockman, R. A.; Hughes, D. L. Org. Biomol. Chem. 2005, 3, 73–83. doi:10.1039/b413052a |
| 4. | Galloway, W. R. J. D.; Díaz-Gavilán, M.; Isidro-Llobet, A.; Spring, D. R. Angew. Chem., Int. Ed. 2009, 48, 1194–1196. doi:10.1002/anie.200805452 |
| 5. | Dow, M.; Fisher, M.; James, T.; Marchetti, F.; Nelson, A. Org. Biomol. Chem. 2012, 10, 17–28. doi:10.1039/c1ob06098h |
| 25. | Rejzek, M.; Stockman, R. A.; Hughes, D. L. Org. Biomol. Chem. 2005, 3, 73–83. doi:10.1039/b413052a |
| 21. | Jirgensons, A.; Kauss, V.; Kalvinsh, I.; Gold, M. R. Synthesis 2000, 1709–1712. doi:10.1055/s-2000-8208 |
| 23. | Crabb, T. A.; Newton, R. F.; Jackson, D. Chem. Rev. 1971, 71, 109–126. doi:10.1021/cr60269a005 |
| 24. | Rader, C. P.; Young, R. L., Jr.; Aaron, H. S. J. Org. Chem. 1965, 30, 1536–1539. doi:10.1021/jo01016a048 |
| 19. | Boyer, F.-D.; Hanna, I. Eur. J. Org. Chem. 2006, 471–482. doi:10.1002/ejoc.200500645 |
| 20. | Schwartz, B. D.; McErlean, C. S. P.; Fletcher, M. T.; Mazomenos, B. E.; Konstantopoulou, M. A.; Kitching, W.; De Voss, J. J. Org. Lett. 2005, 7, 1173–1176. doi:10.1021/ol050143w |
| 25. | Rejzek, M.; Stockman, R. A.; Hughes, D. L. Org. Biomol. Chem. 2005, 3, 73–83. doi:10.1039/b413052a |
| 15. | Stockman, R. A. Tetrahedron Lett. 2000, 41, 9163–9165. doi:10.1016/S0040-4039(00)01640-3 |
| 16. | Stockman, R. A.; Sinclair, A.; Arini, L. G.; Szeto, P.; Hughes, D. L. J. Org. Chem. 2004, 69, 1598–1602. doi:10.1021/jo035639a |
| 17. | Rejzek, M.; Stockman, R. A.; van Maarseveen, J. H.; Hughes, D. L. Chem. Commun. 2005, 4661–4662. doi:10.1039/b508969g |
| 18. | Robbins, D.; Newton, A. F.; Gignoux, C.; Legeay, J.-C.; Sinclair, A.; Rejzek, M.; Laxon, C. A.; Yalamanchili, S. K.; Lewis, W.; O’Connell, M. A.; Stockman, R. A. Chem. Sci. 2011, 2, 2232–2235. doi:10.1039/c1sc00371b |
| 14. | Happ, G. M.; Eisner, T. Science 1961, 134, 329–331. doi:10.1126/science.134.3475.329 |
| 22. | Scarpi, D.; Occhiato, E. G.; Guarna, A. J. Org. Chem. 1999, 64, 1727–1732. doi:10.1021/jo981946i |
© 2012 O’Connell et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)