Abstract
We present a mechanistic investigation of Pd-catalyzed allylic substitution of cyclic gem-difluorinated carbonates 1 and 4, previously employed in the synthesis of 3',3'-difluoro-2'-hydroxymethyl-4',5'-unsaturated carbocyclic nucleosides in 17 steps. The substitution features a reversal of regioselectivity caused by fluorine.

Graphical Abstract
Background
Carbocyclic nucleosides (CNAs), in which the furanose oxygen atoms of the 4'-oxonucleosides are substituted by CH2, have received considerable attention because they exhibit greater metabolic stability toward nucleoside phosphorylases and higher lipophilicity, two properties that are potentially beneficial in terms of increased in vivo half life, oral efficiency and cell wall penetration [1,2]. Based on CNA skeletons, 1,2-disubstituted carbocyclic nucleosides (OTCs) recently attracted more and more attention [3-9], especially after De Clercq et al. found that some OTCs showed moderate to good activity against murine leukemia cells L1210/0, human T-lymphocyte cells Molt4/C8, and CEM/0 via topological substructural approach to molecular design (TOSS-MODE) [10]. As part of our ongoing and continual efforts to prepare potential bioactive fluorinated nucleosides, our group recently described the stereoselective synthesis of 3',3'-difluoro-4',5'-unsaturated OTCs 2–3 and 5 [11]. The whole synthesis highlighted the stereoselective Reformatskii-Claisen rearrangement, ring-closing metathesis (RCM), and Pd-catalyzed allylic substitution, in which the regioselectivity was reversed from that of nonfluorinated substrates. This reversed regioselectivity caused by fluorine interests us greatly. Herein, we present a mechanistic investigation of Pd-catalyzed allylic substitution of cyclic gem-difluorinated carbonates.
Results and Discussion
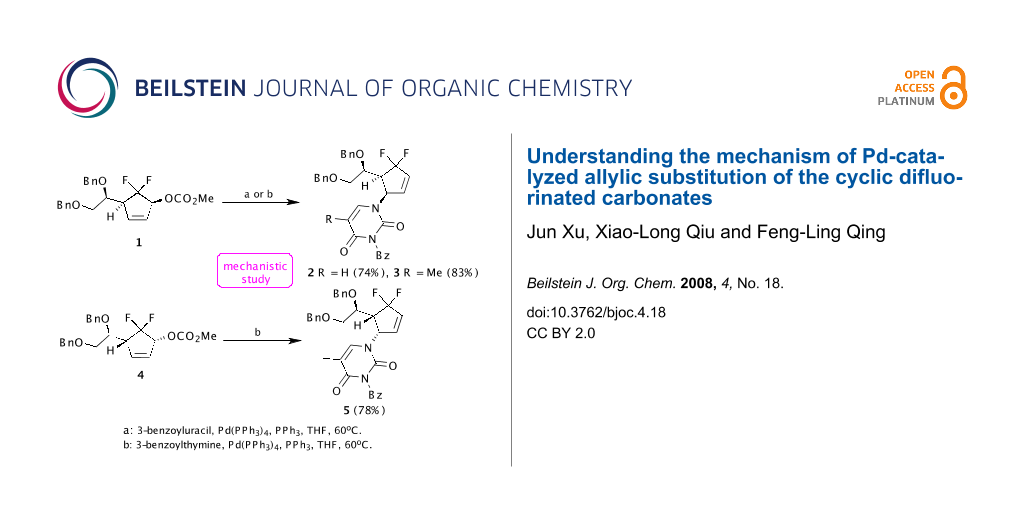
On installation of pyrimidine bases into the gem-difluorinated allylic carbonates 1 and 4, our group found that the γ-substitution products 2, 3 and 5 were surprisingly generated exclusively in good yields, respectively, when the compounds 1 and 4 reacted with suitably protected nucleobases 3-benzoyluracil and 3-benzoylthymine in the presence of a catalytic amount of Pd(PPh3)4 at 60 °C in THF (Scheme 1) [11]. The exclusive regioselectivities of Pd-catalyzed allylic alkylation (Pd-AA) reactions were very interesting. Although Konno et al. have reported that the electron-withdrawing fluoroalkyl groups would alter the regioselectivities of acyclic allylic alkylation compared with their non-fluorinated counterparts [12-17], their reactions mostly concerned the Pd-catalyzed regio- and stereoselective formate reduction of fluorine-containing allylic mesylates. To the best of our knowledge, the effect of gem-difluoromethylene group on Pd-catalyzed cyclic allylic substitution has never been addressed so far. The regioselectivity was totally different from those of nonfluorinated substrates [18].
![[1860-5397-4-18-i1]](/bjoc/content/inline/1860-5397-4-18-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pd-catalyzed allylic substitution of the gem-difluorinated allylic carbonates 1 and 4.
Scheme 1: Pd-catalyzed allylic substitution of the gem-difluorinated allylic carbonates 1 and 4.
Unexpected and specific regioselectivity of Pd-catalytic asymmetric reactions of the gem-difluorinated allylic intermediates 1 and 4 prompted us to investigate further the mechanism of these reactions. Currently, one of the most direct tactics for mechanistic investigation of Pd-AA reaction was built on the analysis of crystal structure and 13C NMR spectroscopy of Pd-π-allyl complex [19,20]. The orientation of attack of nucleophiles on the Pd-π-allyl complex could be illustrated via examining the 13C NMR chemical shifts of three carbon atoms attached to the palladium. According to the model of DeShong et al. [21], it was anticipated that a symmetrical Pd-π-allyl complex should be temporarily generated once the compounds 1 or 4 were treated with palladium catalyst. Thus, α-substitution products should be afforded considering the steric effects. However, only γ-substitution products 2–3 and 5 were isolated in our case, which, in our opinion, resulted from the specific electron-withdrawing property of the gem-difluoromethylene group. To further validate our hypothesis and the proposed model of DeShong et al., we decided to explore the crystal structure and 13C NMR of the corresponding Pd-π-allyl complex.
In 2000, Bäckvall and co-workers investigated the X-ray structures for cis and trans isomers of [(1,2,3-η)-4-acetoxycyclohex-2-enyl]palladium chloride dimers [22]. They found that the X-ray structure of trans-trans dimer displayed asymmetric allyl-palladium bonding where the Pd-C3 bond was shorter than the Pd-C1 bond. Their study provided the first direct evidence for the presence of electronic interaction in the Pd-catalytic asymmetric allylic reactions of 1-acetoxy-4-chloro-2-cyclohexene. Based on their study, we first converted the precursor compound 6 to the γ-chloro-subsituted compound 7 in 78% yield by treatment with NCS / PPh3 in THF at 0 °C (Scheme 2). Using the stereodivergent method of Kurosawa et al. [23], trans-trans dimer 8 and cis-cis dimer 9 were prepared in 74% and 86% yield by treatment of the chloride 7 with Pd(dba)2 using toluene and DMSO as the solvent, respectively.
![[1860-5397-4-18-i2]](/bjoc/content/inline/1860-5397-4-18-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of trans-trans dimer 8 and syn-syn dimer 9.
Scheme 2: Synthesis of trans-trans dimer 8 and syn-syn dimer 9.
To our delight, the crystal of trans-trans dimer 8 was suitable for X-ray analysis (Figure 1). It was clear that 1,2-bis(benzyloxy)ethyl moieties in dimer 8 occupied the positions trans to the corresponding palladium atoms. Also obvious was that allyl-palladium bonding in trans-trans dimer 8 was almost symmetric: within experimental error, there was not much difference between the Pd1-C2 bond length 2.105 Å (11) and Pd1-C4 bond length 2.118 Å (9), and between the Pd1-C4-C3 bond angle 68.9° (6) and Pd1-C2-C3 bond angle 69.2° (7) (Table 1). Thus, as expected from the proposed model [21], the symmetrical Pd-π-allyl complex was generated. According to the X-ray structure of the trans-trans dimer 8, it was clear that C4 position was more shielded than the C2 position, which should guide the attack of nucleophiles from the C2 position. However, we only isolated the product resulting from the attack of nucleophiles from C4 position, which, in our opinion, was totally attributed to the strong electron-withdrawing property of neighboring gem-difluoromethylene group. 13C NMR spectroscopy of the trans-trans dimer 8 unambiguously demonstrated that C2 (t, δ = 73.2) experienced higher field than C4 (s, δ = 81.4), which was also ascribed to the strong electron-withdrawing property of the CF2 group. Taking all the above analysis together, it seems that the CF2 group's strong electron-withdrawing property leads to a higher density of positive charge at the C4 position of the π-allyl palladium, which predominantly promotes attack of the nucleophile on C4 even when the C4 position is more hindered than the C2 position.
![[1860-5397-4-18-1]](/bjoc/content/figures/1860-5397-4-18-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray structure of trans-trans dimer 8.
Figure 1: X-ray structure of trans-trans dimer 8.
Table 1: Selected bond lengths (Å), bond angle (o) and chemical shifts of 13C NMR spectroscopy of the trans-trans dimer 8.
| bond length | bond angle | 13C NMR | |
|---|---|---|---|
| Pd1-C2 | 2.105 Å (11) | ––––– | ––––– |
| Pd1-C4 | 2.118 Å (9) | ––––– | ––––– |
| Pd1-C4-C3 | ––––– | 68.9° (6) | ––––– |
| Pd1-C2-C3 | ––––– | 69.2° (7) | ––––– |
| C2 | ––––– | ––––– | 73.1 (t, J = 19.3 Hz) |
| C4 | ––––– | ––––– | 81.4 (s) |
Interestingly, we also found that treatment of the trans-trans dimer 8 with PPh3 / 3-benzoylthymine sodium at rt provided the nucleoside analogue 5 in 30% yield. No product was detected when PPh3 was absent (Scheme 3). Thus, we think that the reaction occurred involving the monopalladium complex 10. The complex 10 resulted from the depolymerization of the dimer 8 caused by PPh3. With this thought in mind, we attempted to isolate and characterize the monopalladium complex. Unfortunately, we found that exposure of the trans-trans dimer 8 to PPh3 / AgSbF6 afforded the monopalladium complex 11, which was too unstable to be isolated (Scheme 4). Neither did substitution of PPh3 by dppe deliver our desired complex 12 but gave instead the palladium complex 13, whose structure was confirmed by X-ray structure analysis. In our opinion, the reason we failed to isolate the monopalladium complex was also due to the presence of gem-difluoromethylene group. That is, in the absence of nucleophiles, the dimer 8 tended to lose the cyclopentanyl ligand upon treatment with PPh3 or dppe, because the CF2 group made the Pd-π-allyl too electron-deficient. That was why we could isolate the palladium complex 13 as the only product.
![[1860-5397-4-18-i3]](/bjoc/content/inline/1860-5397-4-18-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of trans-trans dimer 8 with 3-benzoylthymine sodium / PPh3.
Scheme 3: Reaction of trans-trans dimer 8 with 3-benzoylthymine sodium / PPh3.
![[1860-5397-4-18-i4]](/bjoc/content/inline/1860-5397-4-18-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of trans-trans dimer 8 with PPh3 / AgSbF6.
Scheme 4: Reaction of trans-trans dimer 8 with PPh3 / AgSbF6.
In conclusion, we have investigated the reaction mechanism of Pd-catalyzed allylic substitution of cyclic gem-difluorinated intermediates in detail via the crystal structure and 13C NMR spectroscopy of the Pd-π-allyl complex. We found that the Pd-catalyzed reactions of cyclic gem-difluorinated allylic carbonates 1 and 4 proceeded via the symmetric Pd-π-allyl bonding and highly regioselective γ-substitution resulted from the neighboring gem-difluoromethylene group. We propose that the present work opens a new avenue for the further insight into the Pd-catalyzed allylic substitution reactions.
Supporting Information
| Supporting Information File 1: Experimental Section and Characterization Data of Compounds | ||
| Format: DOC | Size: 6.2 MB | Download |
References
-
Marquez, V. E. Adv. Antiviral Drug Des. 1996, 2, 89–146. doi:10.1016/S1075-8593(96)80104-3
Return to citation in text: [1] -
Zhu, X.-F. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 651–690. doi:10.1080/15257770008035015
Return to citation in text: [1] -
Santana, L.; Teijeira, M.; Uriarte, E.; Terán, C.; Casellato, U.; Graziani, R. Nucleosides Nucleotides 1996, 15, 1179–1187. doi:10.1080/07328319608007386
Return to citation in text: [1] -
Escuredo, V.; Ferro, B.; Santana, L.; Teijeira, M.; Uriarte, E. Nucleosides Nucleotides 1997, 16, 1453–1456. doi:10.1080/07328319708006204
Return to citation in text: [1] -
Besada, P.; Terán, C.; Teijeira, M.; Uriarte, E. Nucleosides Nucleotides 1999, 18, 725–726. doi:10.1080/15257779908041554
Return to citation in text: [1] -
Terán, C.; González-Moa, M. J.; Mosquera, R.; Santana, L. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 999–1002. doi:10.1081/NCN-100002477
Return to citation in text: [1] -
Santana, L.; Teijeira, M.; Terán, C.; Uriarte, E.; Viña, D. Synthesis 2001, 1532–1538. doi:10.1055/s-2001-16075
Return to citation in text: [1] -
Besada, P.; González-Moa, M. J.; Terán, C.; Santana, L.; Uriarte, E. Synthesis 2002, 2445–2449. doi:10.1055/s-2002-35239
Return to citation in text: [1] -
González-Moa, M. J.; Besada, P.; Teijeira, M.; Terán, C.; Uriarte, E. Synthesis 2004, 543–548. doi:10.1055/s-2004-815975
Return to citation in text: [1] -
Estrada, E.; Uriarte, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. J. Med. Chem. 2000, 43, 1975–1985. doi:10.1021/jm991172d
Return to citation in text: [1] -
Yang, Y.-Y.; Xu, J.; You, Z.-W.; Xu, X.-H.; Qiu, X.-L.; Qing, F.-L. Org. Lett. 2007, 9, 5437–5440. doi:10.1021/ol7023955
Return to citation in text: [1] [2] -
Konno, T.; Ishihara, T.; Yamanaka, H. Tetrahedron Lett. 2000, 41, 8467–8472. doi:10.1016/S0040-4039(00)01485-4
Return to citation in text: [1] -
Konno, T.; Daitoh, T.; Ishihara, T.; Yamanaka, H. Tetrahedron: Asymmetry 2001, 12, 2743–2748. doi:10.1016/S0957-4166(01)00485-2
Return to citation in text: [1] -
Okano, T.; Matsubara, H.; Kusukawa, T.; Fujita, M. J. Organomet. Chem. 2003, 676, 43–48. doi:10.1016/S0022-328X(03)00262-6
Return to citation in text: [1] -
Konno, T.; Takehana, T.; Ishihara, T.; Yamanaka, H. Org. Biomol. Chem. 2004, 2, 93–98. doi:10.1039/b310950j
Return to citation in text: [1] -
Konno, T.; Nagata, K.; Ishihara, T.; Yamanaka, H. J. Org. Chem. 2002, 67, 1768–1775. doi:10.1021/jo011013d
Return to citation in text: [1] -
Konno, T.; Takehana, T.; Mishima, M.; Ishihara, T. J. Org. Chem. 2006, 71, 3545–3550. doi:10.1021/jo0602120
Return to citation in text: [1] -
Nokami, J.; Matsuura, H.; Nakasima, K.; Shibata, S. Chem. Lett. 1994, 23, 1071–1074. doi:10.1246/cl.1994.1071
Return to citation in text: [1] -
Szabó, K. J. Chem. Soc. Rev. 2001, 30, 136–143. doi:10.1039/a908934i
Return to citation in text: [1] -
Macsári, I.; Hupe, E.; Szabó, K. J. J. Org. Chem. 1999, 64, 9547–9556. doi:10.1021/jo991172l
Return to citation in text: [1] -
Hoke, M. E.; Brescia, M.-R.; Bogaczyk, S.; DeShong, P.; King, B. W.; Crimmins, M. T. J. Org. Chem. 2002, 67, 327–335. doi:10.1021/jo010672n
Return to citation in text: [1] [2] -
Jonasson, C.; Kritikos, M.; Bäckvall, J.-E.; Szabó, K. J. Chem.–Eur. J. 2000, 6, 432–436. doi:10.1002/(SICI)1521-3765(20000204)6:3<432::AID-CHEM432>3.0.CO;2-5
Return to citation in text: [1] -
Kurosawa, H.; Ogoshi, S.; Kawasaki, Y.; Murai, S.; Miyoshi, M.; Ikeda, I. J. J. Am. Chem. Soc. 1990, 112, 2813–2814. doi:10.1021/ja00163a060
Return to citation in text: [1]
| 1. | Marquez, V. E. Adv. Antiviral Drug Des. 1996, 2, 89–146. doi:10.1016/S1075-8593(96)80104-3 |
| 2. | Zhu, X.-F. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 651–690. doi:10.1080/15257770008035015 |
| 11. | Yang, Y.-Y.; Xu, J.; You, Z.-W.; Xu, X.-H.; Qiu, X.-L.; Qing, F.-L. Org. Lett. 2007, 9, 5437–5440. doi:10.1021/ol7023955 |
| 11. | Yang, Y.-Y.; Xu, J.; You, Z.-W.; Xu, X.-H.; Qiu, X.-L.; Qing, F.-L. Org. Lett. 2007, 9, 5437–5440. doi:10.1021/ol7023955 |
| 10. | Estrada, E.; Uriarte, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. J. Med. Chem. 2000, 43, 1975–1985. doi:10.1021/jm991172d |
| 3. | Santana, L.; Teijeira, M.; Uriarte, E.; Terán, C.; Casellato, U.; Graziani, R. Nucleosides Nucleotides 1996, 15, 1179–1187. doi:10.1080/07328319608007386 |
| 4. | Escuredo, V.; Ferro, B.; Santana, L.; Teijeira, M.; Uriarte, E. Nucleosides Nucleotides 1997, 16, 1453–1456. doi:10.1080/07328319708006204 |
| 5. | Besada, P.; Terán, C.; Teijeira, M.; Uriarte, E. Nucleosides Nucleotides 1999, 18, 725–726. doi:10.1080/15257779908041554 |
| 6. | Terán, C.; González-Moa, M. J.; Mosquera, R.; Santana, L. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 999–1002. doi:10.1081/NCN-100002477 |
| 7. | Santana, L.; Teijeira, M.; Terán, C.; Uriarte, E.; Viña, D. Synthesis 2001, 1532–1538. doi:10.1055/s-2001-16075 |
| 8. | Besada, P.; González-Moa, M. J.; Terán, C.; Santana, L.; Uriarte, E. Synthesis 2002, 2445–2449. doi:10.1055/s-2002-35239 |
| 9. | González-Moa, M. J.; Besada, P.; Teijeira, M.; Terán, C.; Uriarte, E. Synthesis 2004, 543–548. doi:10.1055/s-2004-815975 |
| 21. | Hoke, M. E.; Brescia, M.-R.; Bogaczyk, S.; DeShong, P.; King, B. W.; Crimmins, M. T. J. Org. Chem. 2002, 67, 327–335. doi:10.1021/jo010672n |
| 23. | Kurosawa, H.; Ogoshi, S.; Kawasaki, Y.; Murai, S.; Miyoshi, M.; Ikeda, I. J. J. Am. Chem. Soc. 1990, 112, 2813–2814. doi:10.1021/ja00163a060 |
| 19. | Szabó, K. J. Chem. Soc. Rev. 2001, 30, 136–143. doi:10.1039/a908934i |
| 20. | Macsári, I.; Hupe, E.; Szabó, K. J. J. Org. Chem. 1999, 64, 9547–9556. doi:10.1021/jo991172l |
| 21. | Hoke, M. E.; Brescia, M.-R.; Bogaczyk, S.; DeShong, P.; King, B. W.; Crimmins, M. T. J. Org. Chem. 2002, 67, 327–335. doi:10.1021/jo010672n |
| 18. | Nokami, J.; Matsuura, H.; Nakasima, K.; Shibata, S. Chem. Lett. 1994, 23, 1071–1074. doi:10.1246/cl.1994.1071 |
| 12. | Konno, T.; Ishihara, T.; Yamanaka, H. Tetrahedron Lett. 2000, 41, 8467–8472. doi:10.1016/S0040-4039(00)01485-4 |
| 13. | Konno, T.; Daitoh, T.; Ishihara, T.; Yamanaka, H. Tetrahedron: Asymmetry 2001, 12, 2743–2748. doi:10.1016/S0957-4166(01)00485-2 |
| 14. | Okano, T.; Matsubara, H.; Kusukawa, T.; Fujita, M. J. Organomet. Chem. 2003, 676, 43–48. doi:10.1016/S0022-328X(03)00262-6 |
| 15. | Konno, T.; Takehana, T.; Ishihara, T.; Yamanaka, H. Org. Biomol. Chem. 2004, 2, 93–98. doi:10.1039/b310950j |
| 16. | Konno, T.; Nagata, K.; Ishihara, T.; Yamanaka, H. J. Org. Chem. 2002, 67, 1768–1775. doi:10.1021/jo011013d |
| 17. | Konno, T.; Takehana, T.; Mishima, M.; Ishihara, T. J. Org. Chem. 2006, 71, 3545–3550. doi:10.1021/jo0602120 |
| 22. | Jonasson, C.; Kritikos, M.; Bäckvall, J.-E.; Szabó, K. J. Chem.–Eur. J. 2000, 6, 432–436. doi:10.1002/(SICI)1521-3765(20000204)6:3<432::AID-CHEM432>3.0.CO;2-5 |
© 2008 Xu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)