Abstract
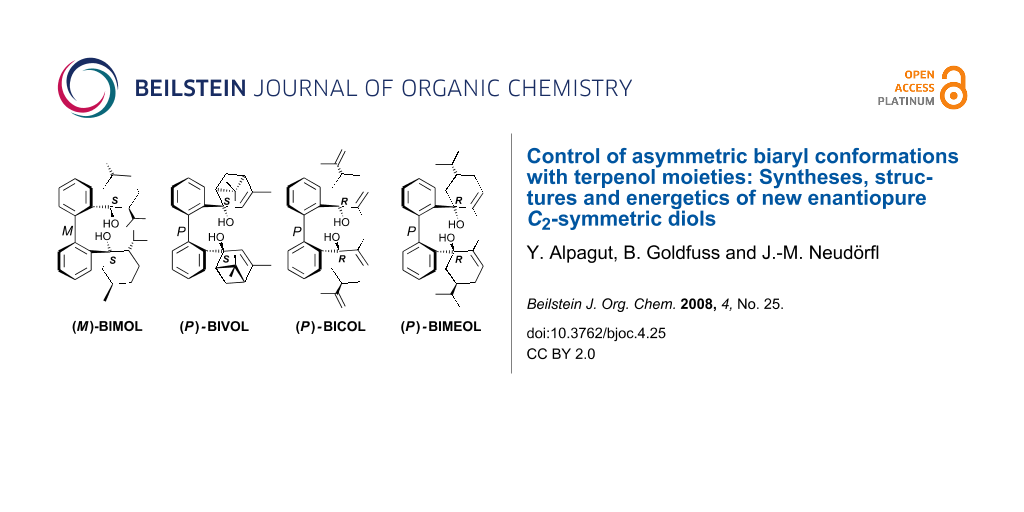
New enantiopure, C2-symmetric biphenyl-2,2′-diols based on (−)-menthone (BIMOL), (−)-verbenone (BIVOL) and (−)-carvone (BICOL and hydrogenated BIMEOL), are accessible via short, synthetic routes. All diols form intramolecular hydrogen bonds and hence can be employed as chelating ligands for catalyst design, as it demonstrated for the (−)-fenchone based BIFOL. The sense of asymmetry of the biphenyl axes is controlled by the chiral terpene units and is conformationally surprisingly stable. X-ray analyses reveal M biphenyl conformation for BIMOL and P biphenyl conformation for each of BIVOL, BICOL and BIMEOL. The origins of the conformational biphenyl preferences are confirmed by computational ONIOM evaluations of the diols and their diastereomeric conformers. The experimentally observed biphenyl conformations are all energetically preferred, i.e. with 1.3 kcal/mol for (M)-BIMOL, with 5.1 kcal/mol for (P)-BIVOL, with 5.8 kcal/mol for (P)-BICOL, and with 5.4 kcal/mol for (P)-BIMEOL.

Graphical Abstract
Introduction
Enantiopure biaryl systems with flexible chiral axes are widespread, e.g. in pharmacological natural products or as ligands in enantioselective catalyses [1]. Chelating C2-symmetric diols such as BINOLs [2-4] and TADDOLs [5,6] are often employed as chiral ligands in enantioselective synthesis. We recently reported the synthesis and the X-ray crystal structure of (M)-BIFOL [7] (biphenyl-2,2′-bisfenchol, Scheme 1) and its derivatives [8-11]. (M)-BIFOL exhibits, in a similar way as BINOLs, a flexible biaryl axis with M conformation, induced by the hydrogen bonded fenchol moieties, and sterically crowded aliphatic alcohol functions, as in TADDOLs. The fenchol moieties were shown to stabilize the biphenyl via intramolecular hydrogen bonds [7].
![[1860-5397-4-25-i1]](/bjoc/content/inline/1860-5397-4-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Atropisomeric BINOL and conformationally restricted, (−)-fenchone-based (M)-BIFOL.
Scheme 1: Atropisomeric BINOL and conformationally restricted, (−)-fenchone-based (M)-BIFOL.
Modular fencholates [12,13] were employed as chiral, chelating ligands, e.g. in enantioselective organozinc catalysts [14-18], in chiral n-butyllithium aggregates [19-23] and in enantioselective Pd- and Cu-catalyzed C-C-couplings [9,10,24]. Here we present syntheses and characterizations of new enantiopure C2-symmetric diols based on (−)-menthone, (−)-verbenone and (−)-carvone, and reveal origins of their conformational biphenyl restrictions.
Results and Discussion
The new 1,1′-biphenyl-2,2′-bisterpenols BIMOL, BIVOL and BICOL were synthesized by addition of 2,2′-dilithiobiphenyl to (−)-menthone, (−)-verbenone and (−)-carvone and subsequent hydrolysis (Scheme 2). The nucleophilic 2,2′-dilithiobiphenyl adds to the carbonyl groups preferably from the sterically less hindered side, i.e. trans to the isopropyl group in (−)-menthone yielding BIMOL, and trans to the isopropenyl group in (−)-carvone [25-27] yielding BICOL. For (−)-verbenone the sterically less crowded side of the pinene backbone, trans to the geminal dimethyl unit, is preferred yielding BIVOL. Catalytic partial hydrogenation of BICOL [28,29] yields the isopropenyl group in BIMEOL retaining the cyclohexene unit (Scheme 2).
![[1860-5397-4-25-i2]](/bjoc/content/inline/1860-5397-4-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of (M)-BIMOL, (P)-BIVOL, (P)-BICOL, and (P)-BIMEOL.
Scheme 2: Syntheses of (M)-BIMOL, (P)-BIVOL, (P)-BICOL, and (P)-BIMEOL.
Intramolecular hydrogen bonds between the hydroxy groups of the terpenol moieties are apparent in the X-ray crystal structures of BIMOL, BIVOL, BICOL and BIMEOL (Figure 1 – Figure 4). These chelating hydroxyl groups enable applications in enantioselective catalysts [9,24] and reagents [8], as has been described for the chelating BIFOL with 2.2 Å for O-H-OH and 3.0 Å for O-O [7]. BIMOL exhibits distances of 1.98 Å for O-H-OH and of 2.80 Å for O-O (Figure 1, Table 1). In the crystal structure of BIVOL, the hydrogen atoms of the hydroxy groups are disordered, but the close distance of 2.71 Å for O-O points also to hydrogen bonding (Figure 2, Table 1). BICOL shows distances of 1.76 Å (O-H-OH) and 2.65 Å (O-O, Figure 3, Table 2) and BIMEOL of 1.67 Å (O-H-OH) and 2.62 Å (O-O, Figure 4, Table 2). In close analogy to the conformationally restricted (M)-BIFOL (Scheme 1), all biphenyl axes of these chelating diols exhibit according to X-ray crystal analyses preferred biphenyl conformations: a M-(R)-sense is found for BIMOL, P-(S)- for BIVOL, P-(S)- for BICOL and likewise P-(S)- for partially hydrogenated BIMEOL (Figure 1–Figure 4, Scheme 2). The C2-C1-C1′-C2′ dihedral angles are −103°, +94°, +96° and +99° respectively (Table 1 and Table 2). Computational ONIOM (B3LYP/6-31++G**:AM1) calculations of the chelating diols prove that the experimentally observed biaryl conformations are indeed intrinsically favored, their alternative diastereomeric biphenyl conformers are all energetically disfavored. (M)-BIMOL is computed to be 1.3 kcal/mol, (P)-BIVOL is 5.1 kcal/mol, (P)-BICOL is 5.8 kcal/mol and (P)-BIMEOL is 5.4 kcal/mol more stable than the conformers (P)-BIMOL, (M)-BIVOL, (M)-BICOL and (M)-BIMEOL (Figure 5 – Figure 12, Table 3). The computationally favored and experimentally found conformers can all form hydrogen bonds between the hydroxy groups, due to favorable arrangements of the terpenol units. The disfavored conformers cannot form such close OH-OH contacts, due to repulsion of the unfavorably aligned bulky terpenol moieties. Donor-solvents like acetone, water or ethanol can bind to the external O-H bonds of the diols (Scheme 3), without disrupting the internal O-H-O bridge, which is crucial for the chiral alignment of the biaryl axis.
![[1860-5397-4-25-1]](/bjoc/content/figures/1860-5397-4-25-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-Ray crystal structure of (M)-BIMOL. An acetone molecule, binding to the external OH group, is omitted for clarity.
Figure 1: X-Ray crystal structure of (M)-BIMOL. An acetone molecule, binding to the external OH group, is omi...
![[1860-5397-4-25-2]](/bjoc/content/figures/1860-5397-4-25-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-Ray crystal structure of (P)-BIVOL. A water molecule, binding to the external OH group, is omitted for clarity (cf. Scheme 3).
Figure 2: X-Ray crystal structure of (P)-BIVOL. A water molecule, binding to the external OH group, is omitte...
![[1860-5397-4-25-3]](/bjoc/content/figures/1860-5397-4-25-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-Ray crystal structure of (P)-BICOL. A water molecule, binding to the external OH group, is omitted for clarity (cf. Scheme 3).
Figure 3: X-Ray crystal structure of (P)-BICOL. A water molecule, binding to the external OH group, is omitte...
![[1860-5397-4-25-4]](/bjoc/content/figures/1860-5397-4-25-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-Ray crystal structure of (P)-BIMEOL. An ethanol molecule, binding to the external OH group, is omitted for clarity (cf. Scheme 3)
Figure 4: X-Ray crystal structure of (P)-BIMEOL. An ethanol molecule, binding to the external OH group, is om...
Table 1: Experimental (X-ray crystal structures) and ONIOM(B3LYP/6-31++G**:AM1) computed geometries of BIMOL and BIVOL in P and M biphenyl conformations.a
| BIMOL | BIVOL | |||||
|---|---|---|---|---|---|---|
| MXray | Mcalc. | Pcalc. | PXray | Pcalc. | Mcalc. | |
| O-H-O(H) [ Å ] | 1.98 | 1.973 | 2.954 | - | 1.942 | 2.071 |
| O-O [ Å ] | 2.80 | 2.912 | 3.678 | 2.71 | 2.904 | 2.957 |
| O-H-O [ ° ] | 171.94 | 160.24 | 132.32 | - | 167.31 | 150.11 |
| C2-C1-C1′-C2′ [ ° ] | −103.2 | −102.00 | +95.21 | +93.9 | +99.03 | −91.61 |
| O-CRest-C2-C1 [ ° ] | +33.1 | +35.55 | −4.64 | −39.9 | −38.16 | +108.37 |
| O-C'Rest-C2′-C1′ [ ° ] | +29.6 | +28.78 | +7.48 | −44.7 | −42.53 | +69.70 |
aThe hydroxyl functions were computed by B3LYP, while AM1 was employed for the rest of the structures. Hydrogens were used as link atoms between the layers.
Table 2: Experimental (X-ray crystal structures) and ONIOM(B3LYP/6-31++G**:AM1) computed geometries of BICOL and BIMEOL in P and M biphenyl conformations.a
| BICOL | BIMEOL | |||||
|---|---|---|---|---|---|---|
| PXray | Pcalc. | Mcalc. | PXray | Pcalc. | Mcalc. | |
| O-H-O(H) [ Å ] | 1.76 | 2.028 | 3.666 | 1.67 | 2.028 | 4.055 |
| O-O [ Å ] | 2.65 | 2.967 | 4.242 | 2.62 | 2.966 | 4.501 |
| O-H-O [ ° ] | 169.3 | 160.15 | 120.57 | 168.1 | 160.02 | 111.43 |
| C2-C1-C1′-C2′ [ ° ] | +95.9 | +99.15 | −100.26 | +99.4 | +98.91 | −103.47 |
| O-CRest-C2-C1 [ ° ] | −42.2 | −34.01 | −28.75 | −32.5 | −29.27 | +1.59 |
| O-C'Rest-C2′-C1′ [ ° ] | −30.1 | −29.13 | −10.77 | −35.6 | −34.18 | −40.11 |
aThe hydroxyl functions were computed by B3LYP, while AM1 was employed for the rest of the structures. Hydrogens were used as link atoms between the layers.
![[1860-5397-4-25-5]](/bjoc/content/figures/1860-5397-4-25-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIMOL, Erel. = 0.0 kcal/mol.
Figure 5: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIMOL, Erel. = 0.0 kcal/mol.
![[1860-5397-4-25-6]](/bjoc/content/figures/1860-5397-4-25-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIMOL, Erel. = 1.3 kcal/mol.
Figure 6: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIMOL, Erel. = 1.3 kcal/mol.
![[1860-5397-4-25-7]](/bjoc/content/figures/1860-5397-4-25-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIVOL, Erel. = 5.1 kcal/mol.
Figure 7: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIVOL, Erel. = 5.1 kcal/mol.
![[1860-5397-4-25-8]](/bjoc/content/figures/1860-5397-4-25-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIVOL, Erel. = 0.0 kcal/mol.
Figure 8: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIVOL, Erel. = 0.0 kcal/mol.
![[1860-5397-4-25-9]](/bjoc/content/figures/1860-5397-4-25-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: B3LYP/6-31++G**:AM1 optimized structure of (M)-BICOL, Erel. = 5.8 kcal/mol.
Figure 9: B3LYP/6-31++G**:AM1 optimized structure of (M)-BICOL, Erel. = 5.8 kcal/mol.
![[1860-5397-4-25-10]](/bjoc/content/figures/1860-5397-4-25-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: B3LYP/6-31++G**:AM1 optimized structure of (P)-BICOL, Erel. = 0.0 kcal/mol.
Figure 10: B3LYP/6-31++G**:AM1 optimized structure of (P)-BICOL, Erel. = 0.0 kcal/mol.
![[1860-5397-4-25-11]](/bjoc/content/figures/1860-5397-4-25-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIMEOL, Erel. = 5.4 kcal/mol.
Figure 11: B3LYP/6-31++G**:AM1 optimized structure of (M)-BIMEOL, Erel. = 5.4 kcal/mol.
![[1860-5397-4-25-12]](/bjoc/content/figures/1860-5397-4-25-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIMEOL, Erel. = 0.0 kcal/mol.
Figure 12: B3LYP/6-31++G**:AM1 optimized structure of (P)-BIMEOL, Erel. = 0.0 kcal/mol.
Table 3: Computed absolute and relative energiesa of the biphenyl diols in P and M biphenyl conformations.
| Computed structures | ONIOM (B3LYP/6-31++G**: AM1) | Erel. [kcal/mol] |
|---|---|---|
| (M)-BIMOL | −152.866360 | 0.0 |
| (P)-BIMOL | −152.866159 | +1.3 |
| (M)-BIVOL | −152.625276 | +5.1 |
| (P)-BIVOL | −152.633361 | 0.0 |
| (M)-BICOL | −152.69155 | +5.8 |
| (P)-BICOL | −152.70077 | 0.0 |
| (M)-BIMEOL | −152.781310 | +5.4 |
| (P)-BIMEOL | −152.789859 | 0.0 |
aThe hydroxyl functions were computed with B3LYP/6-31++G**, while AM1 was employed for the rest of the structures. Hydrogen atoms were used as link atoms between the layers.
![[1860-5397-4-25-i3]](/bjoc/content/inline/1860-5397-4-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Co-solvent adducts in the X-ray structures of (M)-BIMOL, (P)-BIVOL, (P)-BICOL, and (P)-BIMEOL.
Scheme 3: Co-solvent adducts in the X-ray structures of (M)-BIMOL, (P)-BIVOL, (P)-BICOL, and (P)-BIMEOL.
Conclusion
The new enantiopure chelating diols (M)-BIMOL, (P)-BIVOL, (P)-BICOL and (P)-BIMEOL are efficiently accessible from coupling of dilithiobiphenyl with (−)-menthone, (−)-verbenone and (−)-carvone. All diols exhibit flexible biaryl axes, which are however conformationally restricted to P or M arrangements. These favored biaryl arrangements are apparent from X-ray analyses and computational comparisons of the biphenyl conformers and arise from suitable hydrogen bonding of the chiral terpenol moieties. As it has been demonstrated for the (−)-fenchone derived (M)-BIFOL, the flexible, but diastereomerically pure, chelating diols can be employed to incorporate many elements or metal ions for the construction of new enantioselective reagents and catalysts.
Experimental
All reactions were carried out under argon atmosphere using Schlenk tube techniques. Solvents were dried by standard methods and distilled under argon prior to use.
Synthesis of Biphenyl-2,2′-bismenthol (M)-BIMOL
2,2′-Dilithiobiphenyl (2 TMEDA) was synthesized by treating biphenyl with n-butyllithium/TMEDA for 24 hours at room temperature [30,31]. To a suspension of 10 g (25.0 mmol) of 2,2′-dilithiobiphenyl (2 TMEDA, M: 398.5 g/mol) in diethyl ether at 0 °C, 7.7 g (8.4 ml, 50.0 mmol) of (−)-menthone were added. The mixture was warmed up to room temperature and was stirred for 24 h. Aqueous work-up followed by 2 recrystallisations from acetone yielded 6.3 g (12.1 mmol, 48%) of (M)-BIMOL. Analytical and spectroscopic data of (M)-BIMOL: mp: 182 °C; calcd: C 83.06, H 10.02, found C 82.97, H 10.04; 1H NMR (CDCl3, 300 MHz) 0.78–0.80 (3H, d), 0.84–0.86 (3H, d), 0.96–0.99 (4H, d), 1.12–1.21 (1H, t), 1.57–1.86 (5H, m), 1.95–2.00 (1H, dd), 2.07–2.12 (1H, d), 2.57 (1H, s), 7.00–7.03 (1H, d), 7.17–7.22 (1H, t), 7.30–7.35 (1H, t), 7.39–7.42 (1H, d); 13C NMR (CDCl3, 75.5 MHz) 144.77, 132.80, 126.75, 125.54, 125.02, 81.16, 47.90, 35.12, 28.17, 26.57, 24.30, 22.25, 21.65, 18.97; [α]20Na = −87.5 (c = 0.4 in n-hexane); EI-MS: 462 (M+), 426 (M+-2H2O); IR (NaCl crystal, cm−1) 3567 (sharp), 3458 (sharp), 3051 (broad), 2946–2860 (sharp). X-Ray crystal data [32] of (M)-BIMOL as clathrate with acetone: C32H46O2* C3H6O, M = 520.79 g/mol; space group P212121; a = 8.9885(5) Å, b = 19.2157(10) Å, c = 36.233(2) Å; V = 6258.2(6) Å3; Z = 8; T = 100(2) K; μ = 0.068 mm−1; reflection total: 31432; unique: 7151; observed: 3612 (I >2σ(I)); parameters refined: 717; R1 = 0.0466, wR2 = 0.0670; GOF = 0.900 (crystallographic data have been deposited with the Cambridge Crystallographic Data Centre).
Synthesis of Biphenyl-2,2′-bisverbenol (P)-BIVOL
To a suspension of 10 g (25.0 mmol) of 2,2′-dilithiobiphenyl (2 TMEDA, M: 398.5g/mol) in diethyl ether at 0 °C, 7.5 g (7.7 ml, 50.0 mmol) of (−)-verbenone were added. The mixture was warmed up to room temperature and was stirred for 24 h. Aqueous work-up followed by 2 recrystallisations from hexane/acetone yielded 4.3 g (6.1 mmol, 24.6 %) of (P)-BIVOL. Analytical and spectroscopic data of (P)-BIVOL: mp: 196 °C; calcd: C 81.99, H 8.65, found C 82.19, H 8.64; 1H NMR (CDCl3, 300 MHz) 0.82 (6H, s), 1.21 (6H, s), 1.31–1.34 (2H, d), 1.66 (2H, s), 2.09–2.20 (4H, m), 2.58 (2H, t), 4.70 (2H, s), 4.76–4.80 (4H, dd), 6.25 (2H, s), 7.23–7.27 (4H, m), 7.31–7.34 (2H, dd), 7.35–7.40 (2H, t); 13C NMR (CDCl3, 75.5 MHz) 151.04, 139.94, 131.41, 128.38, 127.29, 126.76, 125.09, 107.12, 50.68, 49.16, 43.56, 35.60, 34.46, 26.01, 21.89; [α]20Na = −149.3 (c = 0.2 in n-hexane); EI-MS: 454 (M+), 418 (M+-2xOH); IR (NaCl plate, cm−1) 3237 (sharp), 3053 (broad), 2973–2911 (sharp). X-ray crystal data [32] of (P)-BIVOL as clathrate with H2O: C32H38O2*H2O; M=472.66 g/mol; space group P213; a = b = c = 20.029(8) Å; V = 8035(6) Å3; Z = 12; T = 100(2) K; μ = 0.072 mm−1; reflection total: 27084; unique: 5878; observed: 4143 (I >2σ(I)); parameters refined: 320; R1 = 0.0473, wR2 = 0.0941; GOF = 0.981 (crystallographic data have been deposited with the Cambridge Crystallographic Data Centre).
Synthesis of Biphenyl-2,2′-biscarvol (P)-BICOL
To a suspension of 10 g (25.0 mmol) of 2,2′-dilithiobiphenyl (2 TMEDA, M: 398.5 g/mol) in diethylether at 0 °C, 7.50 g (7.8 ml, 50.0 mmol) of (−)-carvone were added. The mixture was warmed up to room temperature and was stirred for 24 h. Aqueous work-up followed by 2 recrystallisations from hexane/ethyl acetate (10:1) yielded 8.4 g (17.8 mmol, 71%) of (P)-BICOL. Analytical and spectroscopic data of (P)-BICOL: mp: 178 °C.; calcd: C 81.32, H 8.53, found C 81.32, H 8.69; 1H NMR (CDCl3, 300 MHz) 1.61 (6H, s), 1.67 (1H, m), 1.97–1.92 (1H, m), 2.24–2.16 (3H, m), 2.46 (1H, s), 4.64–4.58 (2H, d), 5.72 (1H, s), 7.07–7.04 (2H, m), 7.27–7.24 (2H, dd), 7.33–7.28 (1H, m); 13C NMR (CDCl3, 75.5 MHz) 148.81, 142.04, 140.75, 136.94, 132.11, 129.18, 126.29, 126.06, 125.40, 108.90, 79.35, 43.99, 37.48, 31.13, 20.73, 17.99; [α]20Na = −12.45 (c = 0.4 in n-hexane); EI-MS: 454 (M+), 436 (M+-H2O); IR (NaCl crystal, cm−1) 3391 (sharp), 3058 (broad), 2962–2919 (sharp), 1643. X-ray crystal data [32] of (P)-BICOL as clathrate with H2O: C32H38O2*H2O; M = 472.66 g/mol; space group P212121; a = 12.3983(4) Å, b = 12.5961(5) Å, c = 17.5876(7) Å; V = 2746.7(2) Å3; Z = 4; T = 100(2) K; μ = 0.071 mm−1; reflection total: 13337; unique: 3368; observed: 2629 (I >2σ(I)); parameters refined: 380; R1 = 0.0343, wR2 = 0.0659; GOF = 0.960 (crystallographic data have been deposited with the Cambridge Crystallographic Data Centre).
Synthesis of Biphenyl-2,2′-bis-p-menthenol (P)-BIMEOL
To a 0 °C cooled solution of 0.5 g of (P)-BICOL (1.1 mmol) in 30 ml of ethanol a small amount of palladium/C was added. The mixture was placed in an autoclave and was degassed three times with hydrogen. Under 30 bar of H2 the mixture was stirred for 24 hours. Then the reaction mixture was filtered, the solvent was removed in vacuo and the resulting white solid was crystallized in ethanol. Recrystallization (two times) from ethanol yielded 0.43 g (0.85 mmol, 77%) of (P)-BIMEOL. Analytic and spectroscopic data of (P)-BIMEOL: mp: 204 °C; calcd: C 80.91, H 9.59, found C 80.59, H 9.57; 1H NMR (CDCl3, 300 MHz) 0.79–0.81 (6H, d), 1.23–1.28 (2H, t), 1.36–1.41 (3H, m), 1.63 (3H, s), 1.75–1.83 (1H, m), 2.09–2.21 (2H, m), 2.55 (1H, s), 3.70–3.77 (1H, q), 5.72 (1H, s), 7.06–7.09 (2H, m), 7.21–7.26 (2H, dd), 7.26–7.33 (1H, m); 13C NMR (CDCl3, 75.5 MHz) 142.09, 140.87, 137.00, 131.98, 129.34, 126.16, 125.85, 125.79, 79.45, 43.26, 36.43, 32.11, 29.17, 19.85, 19.23, 17.92; [α]20Na = −51.7 (c = 0.4 in n-hexane); EI-MS: 458 (M+), 440 (M+-H2O); IR (NaCl crystal, cm−1) 3349 (sharp), 3046 (broad), 2957-2880 (sharp); X-ray crystal data [32] of (P)-BIMEOL as clathrate with ethanol: C32H42O2*C2H6O; M=504.74 g/mol; space group P212121; a = 11.4105(1) Å, b = 11.9179(2) Å, c = 12.3467(3) Å; V = 3038.91(7) Å3; Z = 4; T = 200(2) K; μ = 0.068 mm−1; reflection total: 25469; unique: 3720; observed: 3028 (I >2σ(I)); parameters refined: 441; R1 = 0.0410, wR2 = 0.1097; GOF = 1.180 (crystallographic data have been deposited with the Cambridge Crystallographic Data Centre).
Computational Section
All computed structures were fully optimized using Morokuma's ONIOM method implemented in GAUSSIAN98 [33]. Hybrid DFT (B3LYP/6-31++G*) levels of theory were applied to the hydroxyl groups, while the rest of the structures were computed by the semiempirical AM1 method. Hydrogen atoms were used as link atoms between the two layers. The structures were analyzed by frequency computations and showed no imaginary frequencies.
References
-
Herrmann, R. Biaryls. In Stereoselective Synthesis; Helmchen, G.; Hoffmann, R. W.; Mulzer, J.; Schaumann, E., Eds.; Methods of Organic Chemistry (Houben-Weyl), Vol. E21f; Thieme: Stuttgart, 1996; pp 5945–5955.
Return to citation in text: [1] -
Brunel, J. M. Chem. Rev. 2005, 105, 857–897. doi:10.1021/cr040079g
Return to citation in text: [1] -
Shibasaki, M.; Sasai, H.; Arai, T. Angew. Chem. 1997, 109, 1290–1311. doi:10.1002/ange.19971091204
Angew. Chem., Int. Ed. Engl. 1997, 36, 1236–1256. doi: 10.1002/anie.199712361.
Return to citation in text: [1] -
Sasai, H.; Arai, T.; Satow, Y.; Houk, K. N.; Shibasaki, M. J. Am. Chem. Soc. 1995, 117, 6194–6198. doi:10.1021/ja00128a005
Return to citation in text: [1] -
Seebach, D.; Beck, A. K.; Heckel, A. Angew. Chem. 2001, 113, 96–142. doi:10.1002/1521-3757(20010105)113:1<96::AID-ANGE96>3.0.CO;2-B
Angew. Chem., Int. Ed. 2001, 40, 92–138. doi: 10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K.
Return to citation in text: [1] -
Braun, M. Angew. Chem. 1996, 108, 565–568. doi:10.1002/ange.19961080508
Angew. Chem., Int. Ed. Engl. 1996, 35, 519. doi: 10.1002/anie.199605191.
Return to citation in text: [1] -
Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7
Return to citation in text: [1] [2] [3] -
Lange, D. A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704–3709. doi:10.1016/j.tet.2006.01.060
Return to citation in text: [1] [2] -
Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6
Return to citation in text: [1] [2] [3] -
Goldfuss, B.; Loschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7
Return to citation in text: [1] [2] -
Soki, F.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2005, 61, 10449–10453. doi:10.1016/j.tet.2005.08.089
Return to citation in text: [1] -
Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7
Return to citation in text: [1] -
Goldfuss, B.; Eisenträger, F. Aust. J. Chem. 2000, 53, 209–212. doi:10.1071/CH99184
Return to citation in text: [1] -
Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211–5218. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785–1792. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166–170. doi:10.1007/s0089400060166
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77–82. doi:10.1021/jo991070v
Return to citation in text: [1] -
Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927–2929. doi:10.1021/om990184u
Return to citation in text: [1] -
Goldfuss, B. Synthesis 2005, 2271–2280. doi:10.1055/s-2005-872107
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem.–Eur. J. 2005, 11, 4019–4023. doi:10.1002/chem.200500158
Return to citation in text: [1] -
Goldfuss, B. Enantioselective addition of organolithiums to C=O groups. In Organo-lithiums in Enantioselective Synthesis; Hodgson, D. M., Ed.; Topics in Organometallic Chemistry, Vol. 5; Springer: Berlin, Germany, 2003; pp 21–36. doi:10.1007/3-540-36117-0_2
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem.–Eur. J. 2001, 7, 4456–4464. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299–4302. doi:10.1002/1521-3757(20001117)112:22<4299::AID-ANGE4299>3.0.CO;2-O
Angew. Chem. Int. Ed. 2000, 112, 4133–4436. doi: 10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X.
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273
Return to citation in text: [1] [2] -
Lecomte, V.; Stéphan, E.; Le Bideau, F.; Jaouen, G. Tetrahedron 2003, 59, 2169–2176. doi:10.1016/S0040-4020(03)00189-3
Return to citation in text: [1] -
Dimitrov, V.; Panev, S. Tetrahedron: Asymmetry 2000, 11, 1513–1516. doi:10.1016/S0957-4166(00)00099-9
Return to citation in text: [1] -
Ashby, E. C.; Laemmle, J. T. Chem. Rev. 1975, 75, 521–546. doi:10.1021/cr60296a005
Return to citation in text: [1] -
Tietze, L. F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischen Praktikum; Georg Thieme Verlag: Stuttgart, New York, 1991; p 265.
Return to citation in text: [1] -
Schneider, D. F.; Viljoen, M. S. Tetrahedron 2002, 58, 5307–5315. doi:10.1016/S0040-4020(02)00492-1
Return to citation in text: [1] -
Neugebauer, W.; Kos, A. J.; Schleyer, P. R. J. Organomet. Chem. 1982, 228, 107–118. doi:10.1016/S0022-328X(00)87089-8
Return to citation in text: [1] -
Schubert, U.; Neugebauer, W.; Schleyer, P. R. J. Chem. Soc., Chem. Commun. 1982, 1184–1185. doi:10.1039/c39820001184
Return to citation in text: [1] -
X-ray analyses were performed by Dr. J.-M. Neudörfl.
Return to citation in text: [1] [2] [3] [4] -
Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.
Return to citation in text: [1]
| 1. | Herrmann, R. Biaryls. In Stereoselective Synthesis; Helmchen, G.; Hoffmann, R. W.; Mulzer, J.; Schaumann, E., Eds.; Methods of Organic Chemistry (Houben-Weyl), Vol. E21f; Thieme: Stuttgart, 1996; pp 5945–5955. |
| 8. | Lange, D. A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704–3709. doi:10.1016/j.tet.2006.01.060 |
| 9. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 10. | Goldfuss, B.; Loschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 11. | Soki, F.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2005, 61, 10449–10453. doi:10.1016/j.tet.2005.08.089 |
| 7. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7 |
| 7. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7 |
| 30. | Neugebauer, W.; Kos, A. J.; Schleyer, P. R. J. Organomet. Chem. 1982, 228, 107–118. doi:10.1016/S0022-328X(00)87089-8 |
| 31. | Schubert, U.; Neugebauer, W.; Schleyer, P. R. J. Chem. Soc., Chem. Commun. 1982, 1184–1185. doi:10.1039/c39820001184 |
| 5. |
Seebach, D.; Beck, A. K.; Heckel, A. Angew. Chem. 2001, 113, 96–142. doi:10.1002/1521-3757(20010105)113:1<96::AID-ANGE96>3.0.CO;2-B
Angew. Chem., Int. Ed. 2001, 40, 92–138. doi: 10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K. |
| 6. |
Braun, M. Angew. Chem. 1996, 108, 565–568. doi:10.1002/ange.19961080508
Angew. Chem., Int. Ed. Engl. 1996, 35, 519. doi: 10.1002/anie.199605191. |
| 9. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 24. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273 |
| 2. | Brunel, J. M. Chem. Rev. 2005, 105, 857–897. doi:10.1021/cr040079g |
| 3. |
Shibasaki, M.; Sasai, H.; Arai, T. Angew. Chem. 1997, 109, 1290–1311. doi:10.1002/ange.19971091204
Angew. Chem., Int. Ed. Engl. 1997, 36, 1236–1256. doi: 10.1002/anie.199712361. |
| 4. | Sasai, H.; Arai, T.; Satow, Y.; Houk, K. N.; Shibasaki, M. J. Am. Chem. Soc. 1995, 117, 6194–6198. doi:10.1021/ja00128a005 |
| 8. | Lange, D. A.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2006, 62, 3704–3709. doi:10.1016/j.tet.2006.01.060 |
| 19. | Goldfuss, B. Synthesis 2005, 2271–2280. doi:10.1055/s-2005-872107 |
| 20. | Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem.–Eur. J. 2005, 11, 4019–4023. doi:10.1002/chem.200500158 |
| 21. | Goldfuss, B. Enantioselective addition of organolithiums to C=O groups. In Organo-lithiums in Enantioselective Synthesis; Hodgson, D. M., Ed.; Topics in Organometallic Chemistry, Vol. 5; Springer: Berlin, Germany, 2003; pp 21–36. doi:10.1007/3-540-36117-0_2 |
| 22. | Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem.–Eur. J. 2001, 7, 4456–4464. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S |
| 23. |
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299–4302. doi:10.1002/1521-3757(20001117)112:22<4299::AID-ANGE4299>3.0.CO;2-O
Angew. Chem. Int. Ed. 2000, 112, 4133–4436. doi: 10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X. |
| 25. | Lecomte, V.; Stéphan, E.; Le Bideau, F.; Jaouen, G. Tetrahedron 2003, 59, 2169–2176. doi:10.1016/S0040-4020(03)00189-3 |
| 26. | Dimitrov, V.; Panev, S. Tetrahedron: Asymmetry 2000, 11, 1513–1516. doi:10.1016/S0957-4166(00)00099-9 |
| 27. | Ashby, E. C.; Laemmle, J. T. Chem. Rev. 1975, 75, 521–546. doi:10.1021/cr60296a005 |
| 14. | Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211–5218. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S |
| 15. | Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785–1792. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0 |
| 16. | Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166–170. doi:10.1007/s0089400060166 |
| 17. | Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77–82. doi:10.1021/jo991070v |
| 18. | Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927–2929. doi:10.1021/om990184u |
| 28. | Tietze, L. F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischen Praktikum; Georg Thieme Verlag: Stuttgart, New York, 1991; p 265. |
| 29. | Schneider, D. F.; Viljoen, M. S. Tetrahedron 2002, 58, 5307–5315. doi:10.1016/S0040-4020(02)00492-1 |
| 12. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7 |
| 13. | Goldfuss, B.; Eisenträger, F. Aust. J. Chem. 2000, 53, 209–212. doi:10.1071/CH99184 |
| 7. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881–884. doi:10.1016/S0040-4020(99)01077-7 |
| 9. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 10. | Goldfuss, B.; Loschmann, T.; Kop-Weiershausen, T.; Neudörfl, J.; Rominger, F. Beilstein J. Org. Chem. 2006, 2, No. 7. doi:10.1186/1860-5397-2-7 |
| 24. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273 |
© 2008 Alpagut et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)