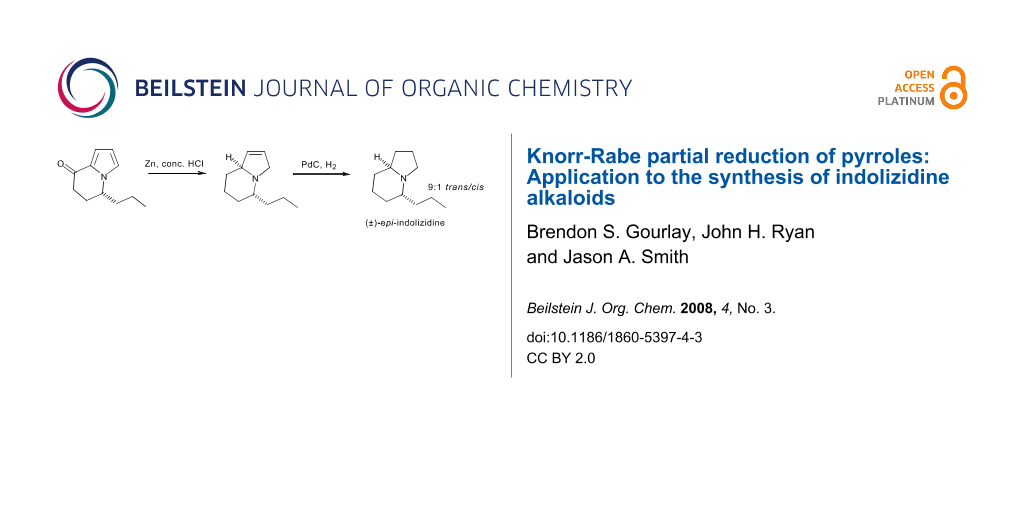
Abstract
Background
The Birch reduction of electron rich pyrroles does not occur readily. However, dissolving metal reduction with zinc under acidic conditions gives 3-pyrrolines (2,5-dihydropyrroles) in reasonable yield. This dissolving metal reduction was first reported by Knorr and Rabe in 1901 but since then has only been reported for the reduction of electron rich pyrroles.
Results
The partial reduction of bicyclic α-ketopyrrole derivatives has been performed under dissolving metal conditions with zinc and hydrochloric acid to give excellent yields of hexahydroindolizidines. This reduction method has been utilised for the diastereoselective synthesis of 5-alkylindolizidines and the stereoselectivity obtained is opposite to that of catalytic hydrogenation.
Conclusion
An efficient stereoselective synthesis of indolizidine alkaloids has been developed from α-ketopyrrole intermediates using a modified version of Knorr and Rabe's pyrrole reduction.

Graphical Abstract
Background
The Birch reaction for the dearomatisation of aromatic substrates is an extremely practical and important tool for synthetic chemists and is used widely as a key step for the synthesis of natural products and molecules of biological interest [1]. However, the partial reduction of pyrrole is difficult as the high electron density of these aromatic heterocycles inhibits the addition of an electron, the first step of a Birch reaction [2]. Donohoe has shown that the partial reduction of pyrroles is possible but this process generally requires the presence of at least two electron withdrawing groups that reduce the electron density of the heterocycle such that reasonable yields of the 3-pyrrolines are obtained [3]. This method was recently exploited for the elegant synthesis of the pyrrolidine alkaloid (±)-1-epiaustraline (3) (Scheme 1) [4].
![[1860-5397-4-3-i1]](/bjoc/content/inline/1860-5397-4-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Donohoe's approach to (±)-1-epiaustraline utilising a modified Birch reduction.
Scheme 1: Donohoe's approach to (±)-1-epiaustraline utilising a modified Birch reduction.
During our studies towards the synthesis of indolizidine alkaloids we required bicyclic 3-pyrrolines and chose to explore accessing these intermediates via partial reduction of the corresponding pyrrole derivatives. These substrates were far more electron rich than those of Donohoe and thus not amenable to Birch reduction methodology. Therefore, we turned to an underutilised reaction that was reported by Knorr and Rabe [5] in 1901 and has only been reported a handful of times since [6-9]. The method employs powdered zinc in an acid media to give 3-pyrrolines, presumably by protonation of the pyrrole to give an iminium ion which is then reduced. It has been shown that reaction of 2,5-dialkylpyrroles gives predominantly the trans 3-pyrroline isomer (Scheme 2) [7-9].
![[1860-5397-4-3-i2]](/bjoc/content/inline/1860-5397-4-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction conditions i) Zn, HCl (aq).
Scheme 2: Reaction conditions i) Zn, HCl (aq).
Results and Discussion
The synthetic plan that we adopted was to construct a bicyclic pyrrole derivative by exploiting the natural reactivity of pyrrole and then to partially reduce the heterocyclic core (Scheme 3). The synthesis started with formation of the γ-pyrrolic ester 7 in high yield using an improved Clauson-Kaas synthesis [10], followed by boron tribromide mediated cyclisation to give the known bicyclic ketone 8 [11]. Upon subjection of this α-ketopyrrole 8 to the modified conditions reported by Andrews and McElvain (slow addition of HCl to the substrate and Zn at 0–10 °C) [5,9] we observed no reaction and starting material was returned. However, when zinc and concentrated HCl were added in small portions to a hot solution of the α-ketopyrrole in methanol over ~10 minutes the starting material was consumed to give the hexahydroindolizidine 9 as the only observable product in ~80% yield. The chemoselectivity using these modified conditions is noteworthy while the carbonyl group is fully reduced the pyrrole group is selectively and partially reduced to the 3-pyrroline. This result was confirmed by comparison of the spectral data with that reported by Huxtable who prepared 9 as an intermediate in the synthesis of lentiginosine [12].
![[1860-5397-4-3-i3]](/bjoc/content/inline/1860-5397-4-3-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction conditions: i) ref. [10] ii) ref. [11] iii) Zn, conc. HCl(aq).
Scheme 3: Reaction conditions: i) ref. [10] ii) ref. [11] iii) Zn, conc. HCl(aq).
For the partial reduction of electron rich pyrroles reported previously, over reduction to give pyrrolidines is a problematic side-reaction. For example, Andrews and McElvain kept the reaction temperature below 10 °C to limit pyrrolidine formation. Under our conditions, starting with the α-ketopyrrole, there was no indication of pyrrolidine formation. The loss of the keto group means that the product is the same as that that would be obtained by reduction of the parent bicyclic pyrrole 13. The reduction of the carbonyl group resembles that of a Clemmensen reduction; however, amalgamated zinc is required for Clemmensen reaction [13].
There are several possible mechanisms for this transformation, however, we propose the first step involves protonation of the carbonyl group to give a conjugated iminium ion 10 (Scheme 4). This species would undergo a two-electron reduction process, with associated protonation to give the α-hydroxy pyrrole 11. Acid-promoted dehydration of 11 would afford a second iminium ion 12 which could undergo further reduction and protonation to give pyrrole 13. The pyrrole could then be protonated to give a third iminium ion 14 and reduction would then give rise to the product 9.
![[1860-5397-4-3-i4]](/bjoc/content/inline/1860-5397-4-3-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Potential mechanism for α-ketopyrrole reduction..
Scheme 4: Potential mechanism for α-ketopyrrole reduction..
Our reaction conditions are much harsher than those previously reported, and yet we do not see pyrrolidine products and this suggests that an alternative pathway is in operation. One possibility is that the intermediate 12 could undergo reduction to give the final product directly without the formation of the pyrrole intermediate 13 (Scheme 5).
![[1860-5397-4-3-i5]](/bjoc/content/inline/1860-5397-4-3-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
To test these hypotheses we reduced the ketone 8 with NaBH4 to give the unstable α-hydroxy pyrrole 11 which was then immediately subjected to the reduction conditions. The same result was obtained giving the 3-pyrroline 9 which lends support to the suggestion that 11 is an intermediate in the reaction. When pyrrole 13 was reacted under the same conditions 9 was formed but the 1H NMR spectrum also showed some starting material remained. The fact that the pyrrole 13 was not observed in the reduction products from α-ketopyrrole 8 lends the support to the suggestion of an alternative pathway. At the present time the intermediacy of 13 cannot be ruled out for the reduction of ketone 8 and alcohol 11.
Due to the facile and rapid reaction of the α-ketopyrrole 8 we explored the potential tandem α-ketopyrrole reduction/catalytic hydrogenation as an alternative to catalytic hydrogenation. The catalytic hydrogenation of 5-substituted tetrahydroindolizidines proceeds with high diastereoselectivity [14,15] and has also been exploited for the synthesis of numerous indolizidine alkaloids [16,17]. The presence of a substituent at C-5 directs the hydrogenation at C-8a from the opposite, least hindered face, to give the cis derivative (Scheme 6).
![[1860-5397-4-3-i6]](/bjoc/content/inline/1860-5397-4-3-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
We were interested in the stereochemical outcome for C-8a using the modified Knorr-Rabe zinc reduction and synthesised the known 5-methyl derivative 18 (Scheme 7) as a model. The methyl ester of (±)-alanine was subjected to the modified Clauson-Kaas pyrrole synthesis to give an α-pyrrolic ester 19 which was subjected to two carbon homologation by ester reduction with DIBAl-H followed by an in situ Wadsworth-Emmons olefination [18]. The alkene 20 was then hydrogenated to the γ-pyrrolic ester 21 and cyclised to give α-ketopyrrole 18 in 67% overall yield from 19. The modified Knorr-Rabe reduction of 18 gave the desired pyrroline 22 in near quantitative yield as a 9:1 mixture of diastereomers. The volatility of the compound meant that for practical purposes it was isolated as the hydrochloride salt by adding concentrated HCl to the organic extract before evaporation. Catalytic hydrogenation of the hydrochloride salt of the pyrroline gave a corresponding mixture of isomers of 5-methylindolizidine 23 but to our surprise the trans isomer was the major diastereomer. The stereochemical assignment of the major and minor isomers was confirmed by comparison of the 13C NMR spectra with the reported spectra for both previously synthesised isomers [19]. The resonance of the carbon signals for C-8a, C-5 and C-3 are diagnostic with these carbons for the major isomer resonating 54.9, 50.2 and 49.1 ppm. This compares to 54.5, 50.0 and 49.2 ppm for the trans isomer and 64.8, 58.9 and 51.8 ppm for the cis isomer as reported in the literature [19]. This result indicates that the major product 22 from the modified Knorr-Rabe zinc reduction has the opposite C-5/C-8a stereochemistry to that typically obtained by catalytic hydrogenation.
![[1860-5397-4-3-i7]](/bjoc/content/inline/1860-5397-4-3-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction conditions: i) DIBAL-H, CH2Cl2, -78 °C, 1 h then triethylphosphonoacetate, NaH, THF, −78 °C – rt ii) H2 (40psi), Pd/C, EtOH iii) BBr3, CH2Cl2, 0 °C, 10 min iv) Zn, conc. HCl(aq) v) H2 (40 psi), Pd/C, EtOH, 2M HCl.
Scheme 7: Reaction conditions: i) DIBAL-H, CH2Cl2, -78 °C, 1 h then triethylphosphonoacetate, NaH, THF, −78 °...
To explain this result we propose that the zinc complexation to the less hindered face of the indolizidine causes protonation to occur on the same side as the C-5 substituent, which results in the trans stereochemistry between C-5 and C-8a.
A beneficial outcome from these observations is that one can now reduce bicyclic intermediates like 18 stereoselectively to enter either diastereomeric series. Corvo has reported the synthesis of the proposed structure of indolizidine 167B by the catalytic hydrogenation of (−)-24 (Scheme 8) [17], and herein we report the racemic synthesis of its epimer (Scheme 9). We have reported the synthesis of the bicyclic ketone (±)-24 [18] and subjection of this α-ketopyrrole to the modified Knorr-Rabe reduction conditions gave the crude 3-pyrroline 26 which was immediately subjected to catalytic hydrogenation to yield a 9:1 mixture of (±)-epi-indolizidine 167B (trans-(±)-27) and (±)-indolizidine 167B (cis-(±)-25) in 91% overall yield from 24. As for the 5-methyl derivative the spectral data of the trans isomer 27 was dramatically different to that of the cis isomer 25 and is consistent with that reported previously [20]. Therefore, this method extends the flexibility of bicyclic pyrroles as intermediates for the synthesis of indolizidine alkaloids, as diastereomeric targets can be accessed simply by the choice of reagent system for reduction of the pyrrole nucleus.
![[1860-5397-4-3-i8]](/bjoc/content/inline/1860-5397-4-3-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-4-3-i9]](/bjoc/content/inline/1860-5397-4-3-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: i) CH3OH, Zn, conc. HCl(aq) ii) H2 (40 psi), Pd/C, EtOH, 2M HCl.
Scheme 9: i) CH3OH, Zn, conc. HCl(aq) ii) H2 (40 psi), Pd/C, EtOH, 2M HCl.
Conclusion
In conclusion, we have discovered a modified method for the Knorr-Rabe partial reduction of electron rich pyrroles which is effective for the reduction of bicyclic α-ketopyrroles to the corresponding 3-pyrroline or hexahydroindolizidine derivatives. The reduction occurs with high diastereoselectivity with 5-alkyl derivatives and gives the opposite diastereoselectivity to that of direct catalytic hydrogenation. This complementary method allows for the synthesis of both diastereomers of indolizidine 167B from a late-stage common intermediate.
Supporting Information
| Supporting Information File 1: Experimental details which includes experimental procedures and spectroscopic data | ||
| Format: DOC | Size: 46.5 KB | Download |
Acknowledgements
The authors thank the University of Tasmania and CSIRO for financial support. The authors would like to thank Peter P. Molesworth of UTAS for a sample of compound 13. JHR thanks University of Tasmania for a Woolmers Lectureship. BSG is grateful for an Australian Postgraduate Award and a CSIRO Postgraduate Top-up Scholarship.
References
-
Subba Rao, G. S. R. Pure Appl. Chem. 2003, 75, 1443–1451. doi:10.1351/pac200375101443
Return to citation in text: [1] -
Donohoe, T. J.; Guyo, P. M. J. Org. Chem. 1996, 61, 7664–7665. doi:10.1021/jo961688u
Return to citation in text: [1] -
Donohoe, T. J.; Guyo, P. M.; Harji, R. R.; Helliwell, M.; Cousins, R. P. C. Tetrahedron Lett. 1998, 39, 3075–3078. doi:10.1016/S0040-4039(98)00362-1
Return to citation in text: [1] -
Donohoe, T. J.; Sintim, H. O. Org. Lett. 2004, 6, 2003–2006. doi:10.1021/ol049397s
Return to citation in text: [1] -
Knorr, L.; Rabe, P.; Bufleb, H.; Jakobi, C. Ber. Dtsch. Chem. Ges. 1901, 34, 3491–3502. doi:10.1002/cber.19010340337
Return to citation in text: [1] [2] -
Schumacher, D. P.; Hall, S. S. J. Am. Chem. Soc. 1982, 104, 6076–6080. doi:10.1021/ja00386a039
Return to citation in text: [1] -
Krawczyk, A. R.; Wróbel, J. T. Pol. J. Chem. 1981, 55, 1363–1367.
Return to citation in text: [1] [2] -
Evans, G. G. J. Am. Chem. Soc. 1951, 73, 5230–5234. doi:10.1021/ja01155a069
Return to citation in text: [1] [2] -
Andrews, L. H.; McElvain, S. M. J. Am. Chem. Soc. 1929, 51, 887–892. doi:10.1021/ja01378a035
Return to citation in text: [1] [2] [3] -
Gourlay, B. S.; Molesworth, P. P.; Ryan, J. H.; Smith, J. A. Tetrahedron Lett. 2006, 47, 799–801. doi:10.1016/j.tetlet.2005.11.104
Return to citation in text: [1] [2] -
Amos, R. I. J.; Gourlay, B. S.; Molesworth, P. P.; Smith, J. A.; Sprod, O. R. Tetrahedron 2005, 61, 8226–8230. doi:10.1016/j.tet.2005.06.026
Return to citation in text: [1] [2] -
Colegate, S. M.; Dorling, P. R.; Huxtable, C. R. Aust. J. Chem. 1984, 37, 1503–1509.
Return to citation in text: [1] -
Clemmensen, E. Ber. Dtsch. Chem. Ges. 1914, 47, 51–63. doi:10.1002/cber.19140470108
Return to citation in text: [1] -
Jefford, C. W.; Tang, Q.; Zaslona, A. J. Am. Chem. Soc. 1991, 113, 3513–3518. doi:10.1021/ja00009a043
Return to citation in text: [1] -
Bond, T. J.; Jenkins, R.; Taylor, P. C. Tetrahedron Lett. 1994, 35, 9263–9266. doi:10.1016/0040-4039(94)88483-8
Return to citation in text: [1] -
Jefford, C. W.; Sienkiewicz, K.; Thornton, S. R. Helv. Chim. Acta 1995, 78, 1511–1524. doi:10.1002/hlca.19950780610
Return to citation in text: [1] -
Corvo, M. C.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X
Return to citation in text: [1] [2] [3] -
Gourlay, B. S.; Little, I.; Ryan, J. H.; Smith, J. A. Nat. Prod. Commun. 2006, 1, 831–837.
Return to citation in text: [1] [2] -
Tehrani, K. A.; D'hooghe, M.; De Kimpe, N. Tetrahedron 2003, 59, 3099–3108. doi:10.1016/S0040-4020(03)00375-2
Return to citation in text: [1] [2] -
Polniaszek, R. P.; Belmont, S. E. J. Org. Chem. 1990, 55, 4688–4693. doi:10.1021/jo00302a038
Return to citation in text: [1]
| 19. | Tehrani, K. A.; D'hooghe, M.; De Kimpe, N. Tetrahedron 2003, 59, 3099–3108. doi:10.1016/S0040-4020(03)00375-2 |
| 18. | Gourlay, B. S.; Little, I.; Ryan, J. H.; Smith, J. A. Nat. Prod. Commun. 2006, 1, 831–837. |
| 19. | Tehrani, K. A.; D'hooghe, M.; De Kimpe, N. Tetrahedron 2003, 59, 3099–3108. doi:10.1016/S0040-4020(03)00375-2 |
| 1. | Subba Rao, G. S. R. Pure Appl. Chem. 2003, 75, 1443–1451. doi:10.1351/pac200375101443 |
| 5. | Knorr, L.; Rabe, P.; Bufleb, H.; Jakobi, C. Ber. Dtsch. Chem. Ges. 1901, 34, 3491–3502. doi:10.1002/cber.19010340337 |
| 14. | Jefford, C. W.; Tang, Q.; Zaslona, A. J. Am. Chem. Soc. 1991, 113, 3513–3518. doi:10.1021/ja00009a043 |
| 15. | Bond, T. J.; Jenkins, R.; Taylor, P. C. Tetrahedron Lett. 1994, 35, 9263–9266. doi:10.1016/0040-4039(94)88483-8 |
| 4. | Donohoe, T. J.; Sintim, H. O. Org. Lett. 2004, 6, 2003–2006. doi:10.1021/ol049397s |
| 16. | Jefford, C. W.; Sienkiewicz, K.; Thornton, S. R. Helv. Chim. Acta 1995, 78, 1511–1524. doi:10.1002/hlca.19950780610 |
| 17. | Corvo, M. C.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X |
| 3. | Donohoe, T. J.; Guyo, P. M.; Harji, R. R.; Helliwell, M.; Cousins, R. P. C. Tetrahedron Lett. 1998, 39, 3075–3078. doi:10.1016/S0040-4039(98)00362-1 |
| 11. | Amos, R. I. J.; Gourlay, B. S.; Molesworth, P. P.; Smith, J. A.; Sprod, O. R. Tetrahedron 2005, 61, 8226–8230. doi:10.1016/j.tet.2005.06.026 |
| 2. | Donohoe, T. J.; Guyo, P. M. J. Org. Chem. 1996, 61, 7664–7665. doi:10.1021/jo961688u |
| 13. | Clemmensen, E. Ber. Dtsch. Chem. Ges. 1914, 47, 51–63. doi:10.1002/cber.19140470108 |
| 11. | Amos, R. I. J.; Gourlay, B. S.; Molesworth, P. P.; Smith, J. A.; Sprod, O. R. Tetrahedron 2005, 61, 8226–8230. doi:10.1016/j.tet.2005.06.026 |
| 12. | Colegate, S. M.; Dorling, P. R.; Huxtable, C. R. Aust. J. Chem. 1984, 37, 1503–1509. |
| 20. | Polniaszek, R. P.; Belmont, S. E. J. Org. Chem. 1990, 55, 4688–4693. doi:10.1021/jo00302a038 |
| 10. | Gourlay, B. S.; Molesworth, P. P.; Ryan, J. H.; Smith, J. A. Tetrahedron Lett. 2006, 47, 799–801. doi:10.1016/j.tetlet.2005.11.104 |
| 10. | Gourlay, B. S.; Molesworth, P. P.; Ryan, J. H.; Smith, J. A. Tetrahedron Lett. 2006, 47, 799–801. doi:10.1016/j.tetlet.2005.11.104 |
| 17. | Corvo, M. C.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X |
| 7. | Krawczyk, A. R.; Wróbel, J. T. Pol. J. Chem. 1981, 55, 1363–1367. |
| 8. | Evans, G. G. J. Am. Chem. Soc. 1951, 73, 5230–5234. doi:10.1021/ja01155a069 |
| 9. | Andrews, L. H.; McElvain, S. M. J. Am. Chem. Soc. 1929, 51, 887–892. doi:10.1021/ja01378a035 |
| 17. | Corvo, M. C.; Pereira, M. M. A. Tetrahedron Lett. 2002, 43, 455–458. doi:10.1016/S0040-4039(01)02189-X |
| 6. | Schumacher, D. P.; Hall, S. S. J. Am. Chem. Soc. 1982, 104, 6076–6080. doi:10.1021/ja00386a039 |
| 7. | Krawczyk, A. R.; Wróbel, J. T. Pol. J. Chem. 1981, 55, 1363–1367. |
| 8. | Evans, G. G. J. Am. Chem. Soc. 1951, 73, 5230–5234. doi:10.1021/ja01155a069 |
| 9. | Andrews, L. H.; McElvain, S. M. J. Am. Chem. Soc. 1929, 51, 887–892. doi:10.1021/ja01378a035 |
| 5. | Knorr, L.; Rabe, P.; Bufleb, H.; Jakobi, C. Ber. Dtsch. Chem. Ges. 1901, 34, 3491–3502. doi:10.1002/cber.19010340337 |
| 9. | Andrews, L. H.; McElvain, S. M. J. Am. Chem. Soc. 1929, 51, 887–892. doi:10.1021/ja01378a035 |
| 18. | Gourlay, B. S.; Little, I.; Ryan, J. H.; Smith, J. A. Nat. Prod. Commun. 2006, 1, 831–837. |
© 2008 Gourlay et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)