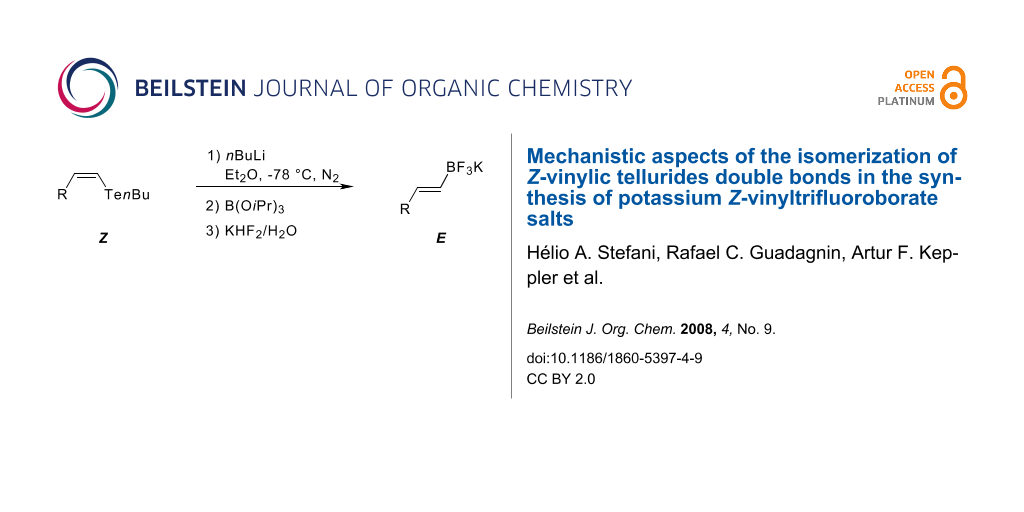
Abstract
Through direct transmetalation reaction of Z-vinylic tellurides with nBuLi was observed the unexpected isomerization of double bonds leading to potassium E-vinyltrifluoroborates salts in low to moderate yields. Using EPR spin trapping experiments the radical species that promoted the stereoinversion of Z-vinylic organometallic species during the preparation of potassium vinyltrifluoroborate salts was identified. The experiments support the proposed mechanism, which is based on the homolytic cleavage of the TenBu bond.

Graphical Abstract
Background
Boronic acids and boronate esters are the most commonly used derivatives in Suzuki-Miyaura cross-coupling reactions. Recently, Molander et al. [1] and our group [2] have explored the use of potassium organotrifluoroborate salts as an alternative to the usual organoboron reagents in alkenyl-alkenyl [3], aryl-aryl [4], alkenyl-alkynyl [5], and alkenyl-aryl [6] cross-coupling reactions.
Distinct from the most commonly explored hydrometallation reactions, the hydrotelluration of alkynes exclusively forms Z-vinylic tellurides [7]. Vinylic tellurides have the ability to undergo tellurium-metal exchange reactions with several different commonly used, commercially available, or easily prepared organometallic reagents, leading to Z-vinyllithiums and Z-vinylcyanocuprates. In reactions promoted by Pd or Ni, these compounds undergo stereospecific coupling with a wide range of organic species [8]. The vinylic organometallic species obtained in this way can also react with carbonyl compounds, α,β-unsaturated systems, or epoxides [9-11] with complete retention of the double-bond stereochemistry.
Taking advantage of the regio- and stereocontrol of the preparation of Z-vinylic tellurides [12], and of the unique features of the transmetallation with complete retention of the original double bond geometry, we report herein the synthesis of potassium vinyltrifluoroborate salts by means of the Te-Li exchange reaction. To the best of our knowledge, this is the first reported preparation of potassium E-vinyltrifluoroborate salts from Z-vinylic tellurides.
Results and Discussion
Functionalized Z-vinylic tellurides 1 were prepared by hydrotelluration of alkynes [13]. Using phenyl vinyl telluride, we performed a series of test reactions to establish the best reaction conditions for the lithium-boron exchange step (Table 1; ii, Scheme 1). Optimum yield was obtained with B(OiPr)3 as the electrophile and ether as the solvent (entry 6).
![[Graphic 1]](/bjoc/content/inline/1860-5397-4-9-i4.svg?max-width=637&scale=1.0)
![[1860-5397-4-9-i1]](/bjoc/content/inline/1860-5397-4-9-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route used to prepare vinyl BF3K salts.
Scheme 1: Synthetic route used to prepare vinyl BF3K salts.
Using the optimized conditions (Table 1, entry 6), all the Z-vinylic tellurides were, to our surprise, transformed into potassium E-vinyltrifluoroborate salts exclusively (see Supporting Information File 1) (Figure 1).
![[1860-5397-4-9-1]](/bjoc/content/figures/1860-5397-4-9-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The 1H NMR spectra of the products showed the presence of the salt nBuBF3K as a by-product (30–50% of the total yield). Use of 1.0 equiv of nBuLi instead of 1.5 equiv as in the optimized protocol gave the same proportion of nBuBF3K.
With 1H NMR, we tried to observe the coupling constants of the vinylic hydrogens for each intermediate of the reaction route. Using this approach, we could prove the cis geometry of the vinylic hydrogens of the intermediate 2 (Scheme 1), which presented a coupling constant of 18.09 Hz [14,15]. Unfortunately, the boronic “ate” complex 4 (Scheme 1) is an insoluble species and no 1H NMR spectra were obtained. However, these results indicated that the double bond geometry isomerization occurred only after the formation of the intermediate 4 (Scheme 1).
We suggest that homolytic cleavage of the Te-Bu bond, from 3 (i, Scheme 1), generates nBu•, which is responsible for the cis-trans isomerization. The butyl radical attack occurs at the boronic “ate” complex 4 (Scheme 1) [16], yielding the nBuBF3K salt as a final product.
In order to verify the presence of radical species in the reaction mixture, we performed EPR spin trapping experiments using 3,5-dibromo-4-nitrosobenzenesulfonate (DBNBS), which is an appropriate spin trap for tellurium centered radicals [17]. Radical species were detected at the i and ii steps of the proposed route. In the first step (i, Scheme 1), the detected spectra contained a mixture of DBNBS radical adducts (Figure 2A). The triplet of triplets (aN = 21.6 G, aH = 0.7 G) is the DBNBS/•TenBu radical adduct [17] and the broadened triplet (aN = 9.1 G, aH = 1.0 G) can be attributed to another DBNBS radical adduct. The intensity of the broadened triplet started to decay after 5 min incubation, and was barely detected in the 15 min incubation spectrum (Figure 2B). The DBNBS/•TenBu signal maintained its intensity during the course of the EPR analysis.
![[1860-5397-4-9-2]](/bjoc/content/figures/1860-5397-4-9-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Representative EPR spectra of DBNBS radical adducts obtained during the Te-Li exchange reaction. (A) EPR spectrum obtained after 1 min incubation of the reaction mixture with the DBNBS aqueous solution, (B) EPR spectrum obtained after 15 min incubation of the reaction mixture with the DBNBS aqueous solution; (○) DBNBS/•TenBu radical adduct and (•) transient DBNBS radical adduct.
Figure 2: Representative EPR spectra of DBNBS radical adducts obtained during the Te-Li exchange reaction. (A...
After the addition of the B(OiPr)3 (ii, Scheme 1), the reaction mixture produced a complex EPR spectra that can be attributed to a mixture of radical species (Figure 3). The addition of the boron reagent generated different radical species from those observed in the previous reaction step (Figure 2).
![[1860-5397-4-9-3]](/bjoc/content/figures/1860-5397-4-9-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Representative EPR spectrum of DBNBS radical adducts obtained during the Li-Boron exchange reaction. EPR spectrum obtained after 15 min incubation of the reaction mixture with the DBNBS aqueous solution.
Figure 3: Representative EPR spectrum of DBNBS radical adducts obtained during the Li-Boron exchange reaction...
We performed control experiments to exclude the possibility of radical generation by the combination of the boron reagent with O2 [18] or by the self-radical generation of the nBuTenBu reagent. Incubation of nBuTenBu, nBuLi and B(OiPr)3 with DBNBS produced no EPR signals (Table 2, entries 3–5). Equimolar solutions of nBuTenBu, nBuLi and DBNBS (Table 2, entry 6) produced a radical signal with similar parameters as those detected during the Te-Li exchange (i, Scheme 1). In the absence of the reducing reagent (nBuLi), an equimolar solution of nBuTenBu, B(OiPr)3 and DBNBS also did not produce EPR signals (Table 2, entry 8).
Table 2: Reactions and Control Experiments Performed.
| Entry | Reactions | DBNBS radical adducts EPR Hyperfines (G) | |||
|---|---|---|---|---|---|
| aN | aH | a′N | a′H | ||
| 1 | BuTeCH=CHPh+nBuLi+DBNBS | 21.6 | 0.7 | 9.1 | 1.0 |
| 2 | LiCH=CHPh+B(OiPr)3+DBNBS | complex signal | |||
| 3 | BuTeBu+DBNBS | no signal | |||
| 4 | nBuLi+DBNBS | no signal | |||
| 5 | B(OiPr)3+DBNBS | no signal | |||
| 6 | BuTeBu+nBuLi+DBNBS | - | - | 9.1 | 1.0 |
| 7 | BuTeBu+nBuLi+B(OiPr)3+DBNBS | complex signal | |||
| 8 | BuTeBu+B(OiPr)3+DBNBS | no signal | |||
To test our proposed mechanism, we repeated the reaction using (Z)-β-bromostyrene, to achieve the desired Z-vinyllithium, the experiments were performed using tBuLi in a solution composed of THF/Et2O/petrol ether, at -120 °C, with and without nBuTenBu, instead of Z-vinylic tellurides to examine the effect of the nBuTenBu as the source of the butyl radical. From this reaction, the expected potassium vinyltrifluoroborate salt was not isolated, probably because it is necessary to use experimental conditions [19] that differ from those that were selected to perform the synthesis of the BF3K salts. To maintain the same reaction conditions, other control experiments were performed (Scheme 2).
![[1860-5397-4-9-i2]](/bjoc/content/inline/1860-5397-4-9-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Experimental conditions. i: 1 equiv nBuLi, Et2O, −78 °C, 30 minutes. ii: 0.8 equiv B(OiPr)3, −20 °C, 60 minutes. iii. 3 equiv KHF2 in aqueous solution, −20 °C to r.t., 30 minutes.
Scheme 2: Experimental conditions. i: 1 equiv nBuLi, Et2O, −78 °C, 30 minutes. ii: 0.8 equiv B(OiPr)3, −20 °C...
Instead of having the double bond isomerization as a radical pathway model, evidence of nBuTenBu radical behavior came from the detection of nBuBF3K as a product only from experiment A (Scheme 2). With the control experiments (Scheme 2), it was proven that the generation of nBuBF3K salt is dependent on the presence of nBuTenBu, as well as that that occurs during the reaction to prepare the alkenyltrifluoroborate salts.
The results presented above support a free radical pathway for the trans-cis double bond isomerization. Scheme 3 was proposed to account for the E-vinyl and nBuBF3K salts. In the first step, the butyl radical 5 is formed by homolytic cleavage of the nBu-Te bond of the compound 3, caused by the lithium species present in the reaction medium. The second step consists of an attack of 5 at the boronic “ate” complex 4, leading to the vinylic radical, which undergoes self-isomerization to the most stable isomer 8. In the third step, the vinylic radical 8 attacks a B(OiPr)3 species, yielding an anionic vinyl boronic “ate” radical. The boron-centered radical is then reduced by a •TenBu radical 6, leading to the E-vinyltrifluoroborate salt 9 after the reaction work up with aqueous KHF2.
![[1860-5397-4-9-i3]](/bjoc/content/inline/1860-5397-4-9-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of the reaction.
Scheme 3: Proposed mechanism of the reaction.
Conclusion
In conclusion, we have identified the radical species that promoted the stereoinversion of vinylic compounds during the preparation of potassium vinyltrifluoroborate salts. The above experiments support the proposed mechanism, which is based on the homolytic cleavage of the TenBu bond.
Supporting Information
| Supporting Information File 1: Experimental section. The file describes the spectral data and the reaction procedure to prepare the potassium vinylorganotrifluoroborate salts | ||
| Format: DOC | Size: 27.5 KB | Download |
References
-
Molander, G. A.; Ellis, N. Acc. Chem. Res. 2007, 40, 275–286. doi:10.1021/ar050199q
Return to citation in text: [1] -
Stefani, H. A.; Cella, R.; Vieira, A. S. Tetrahedron 2007, 63, 3623–3658. doi:10.1016/j.tet.2007.01.061
Return to citation in text: [1] -
Cella, R.; Orfão, A. T. G.; Stefani, H. A. Tetrahedron Lett. 2006, 47, 5075–5078. doi:10.1016/j.tetlet.2006.05.088
Return to citation in text: [1] -
Cella, R.; Cunha, R. L. O. R.; Reis, A. E. S.; Pimenta, D. C.; Klitzke, C. F.; Stefani, H. A. J. Org. Chem. 2006, 71, 244–250. doi:10.1021/jo052061r
Return to citation in text: [1] -
Stefani, H. A.; Cella, R.; Dörr, F. A.; Pereira, C. M. P.; Zeni, G.; Gomes, M., Jr. Tetrahedron Lett. 2005, 46, 563–567. doi:10.1016/j.tetlet.2004.11.160
Return to citation in text: [1] -
Cella, R.; Stefani, H. A. Tetrahedron 2006, 62, 5656–5662. doi:10.1016/j.tet.2006.03.090
Return to citation in text: [1] -
Zeni, G.; Lüdtke, D. S.; Panatieri, R. B.; Braga, A. L. Chem. Rev. 2006, 106, 1032–1076. doi:10.1021/cr0505730
Return to citation in text: [1] -
Zeni, G.; Braga, A. L.; Stefani, H. A. Acc. Chem. Res. 2003, 36, 731–738. doi:10.1021/ar0202621
Return to citation in text: [1] -
Barros, S. M.; Comasseto, J. V.; Berriel, J. N. Tetrahedron Lett. 1989, 30, 7353–7356. doi:10.1016/S0040-4039(00)70695-2
Return to citation in text: [1] -
Hirro, T.; Kambe, N.; Ogawa, A.; Miyoshi, N.; Murai, S.; Sonoda, N. Angew. Chem., Int. Ed. Engl. 1987, 11, 1187–1188. doi:10.1002/anie.198711871
Return to citation in text: [1] -
Marino, J. P.; Tucci, F.; Comasseto, J. V. Synlett 1993, 761–763. doi:10.1055/s-1993-22600
Return to citation in text: [1] -
Barros, S. M.; Dabdoub, M. J.; Dabdoub, V. M. B.; Comasseto, J. V. Organometallics 1989, 8, 1661–1665. doi:10.1021/om00109a014
(experimental procedures).
Return to citation in text: [1] -
Zeni, G.; Formiga, H. B.; Comasseto, J. V. Tetrahedron Lett. 2000, 41, 1311–1313. doi:10.1016/S0040-4039(99)02297-2
Return to citation in text: [1] -
Nesmeyanov, A. N.; Borisov, A. E. Tetrahedron 1957, 1, 158–168. doi:10.1016/0040-4020(57)85018-2
Return to citation in text: [1] -
Seyferth, D.; Vaughan, L. G. J. Am. Chem. Soc. 1964, 86, 883–890. doi:10.1021/ja01059a028
Return to citation in text: [1] -
Pozzi, D.; Scanlan, E. M.; Renaud, P. J. Am. Chem. Soc. 2005, 127, 14204–14205. doi:10.1021/ja055691j
Return to citation in text: [1] -
Keppler, A. F.; Cerchiaro, G.; Augusto, O.; Miyamoto, S.; Prado, F.; Di Mascio, P.; Comasseto, J. V. Organometallics 2006, 25, 5059–5066. doi:10.1021/om060560s
Return to citation in text: [1] [2] -
Cadot, C.; Cossy, J.; Dalko, P. I. Chem. Commun. 2000, 1017–1018. doi:10.1039/b001903h
Return to citation in text: [1] -
Neumann, H.; Seebach, D. Tetrahedron Lett. 1976, 17, 4839–4842. doi:10.1016/S0040-4039(00)78926-X
(experimental details).
Return to citation in text: [1]
| 19. |
Neumann, H.; Seebach, D. Tetrahedron Lett. 1976, 17, 4839–4842. doi:10.1016/S0040-4039(00)78926-X
(experimental details). |
| 1. | Molander, G. A.; Ellis, N. Acc. Chem. Res. 2007, 40, 275–286. doi:10.1021/ar050199q |
| 5. | Stefani, H. A.; Cella, R.; Dörr, F. A.; Pereira, C. M. P.; Zeni, G.; Gomes, M., Jr. Tetrahedron Lett. 2005, 46, 563–567. doi:10.1016/j.tetlet.2004.11.160 |
| 17. | Keppler, A. F.; Cerchiaro, G.; Augusto, O.; Miyamoto, S.; Prado, F.; Di Mascio, P.; Comasseto, J. V. Organometallics 2006, 25, 5059–5066. doi:10.1021/om060560s |
| 4. | Cella, R.; Cunha, R. L. O. R.; Reis, A. E. S.; Pimenta, D. C.; Klitzke, C. F.; Stefani, H. A. J. Org. Chem. 2006, 71, 244–250. doi:10.1021/jo052061r |
| 18. | Cadot, C.; Cossy, J.; Dalko, P. I. Chem. Commun. 2000, 1017–1018. doi:10.1039/b001903h |
| 3. | Cella, R.; Orfão, A. T. G.; Stefani, H. A. Tetrahedron Lett. 2006, 47, 5075–5078. doi:10.1016/j.tetlet.2006.05.088 |
| 16. | Pozzi, D.; Scanlan, E. M.; Renaud, P. J. Am. Chem. Soc. 2005, 127, 14204–14205. doi:10.1021/ja055691j |
| 2. | Stefani, H. A.; Cella, R.; Vieira, A. S. Tetrahedron 2007, 63, 3623–3658. doi:10.1016/j.tet.2007.01.061 |
| 17. | Keppler, A. F.; Cerchiaro, G.; Augusto, O.; Miyamoto, S.; Prado, F.; Di Mascio, P.; Comasseto, J. V. Organometallics 2006, 25, 5059–5066. doi:10.1021/om060560s |
| 9. | Barros, S. M.; Comasseto, J. V.; Berriel, J. N. Tetrahedron Lett. 1989, 30, 7353–7356. doi:10.1016/S0040-4039(00)70695-2 |
| 10. | Hirro, T.; Kambe, N.; Ogawa, A.; Miyoshi, N.; Murai, S.; Sonoda, N. Angew. Chem., Int. Ed. Engl. 1987, 11, 1187–1188. doi:10.1002/anie.198711871 |
| 11. | Marino, J. P.; Tucci, F.; Comasseto, J. V. Synlett 1993, 761–763. doi:10.1055/s-1993-22600 |
| 13. | Zeni, G.; Formiga, H. B.; Comasseto, J. V. Tetrahedron Lett. 2000, 41, 1311–1313. doi:10.1016/S0040-4039(99)02297-2 |
| 8. | Zeni, G.; Braga, A. L.; Stefani, H. A. Acc. Chem. Res. 2003, 36, 731–738. doi:10.1021/ar0202621 |
| 14. | Nesmeyanov, A. N.; Borisov, A. E. Tetrahedron 1957, 1, 158–168. doi:10.1016/0040-4020(57)85018-2 |
| 15. | Seyferth, D.; Vaughan, L. G. J. Am. Chem. Soc. 1964, 86, 883–890. doi:10.1021/ja01059a028 |
| 7. | Zeni, G.; Lüdtke, D. S.; Panatieri, R. B.; Braga, A. L. Chem. Rev. 2006, 106, 1032–1076. doi:10.1021/cr0505730 |
| 6. | Cella, R.; Stefani, H. A. Tetrahedron 2006, 62, 5656–5662. doi:10.1016/j.tet.2006.03.090 |
| 12. |
Barros, S. M.; Dabdoub, M. J.; Dabdoub, V. M. B.; Comasseto, J. V. Organometallics 1989, 8, 1661–1665. doi:10.1021/om00109a014
(experimental procedures). |
© 2008 Stefani et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)