Abstract
Tetrakis(dimethylamino)ethylene (TDAE 1), has been exploited for the first time as a mild reagent for the reduction of arenediazonium salts to aryl radical intermediates through a single electron transfer (SET) pathway. Cyclization of the aryl radicals produced in this way led, in appropriate substrates, to syntheses of indolines and indoles. Cascade radical cyclizations of aryl radicals derived from arenediazonium salts are also reported. The relative ease of removal of the oxidized by-products of TDAE from the reaction mixture makes the methodology synthetically attractive.

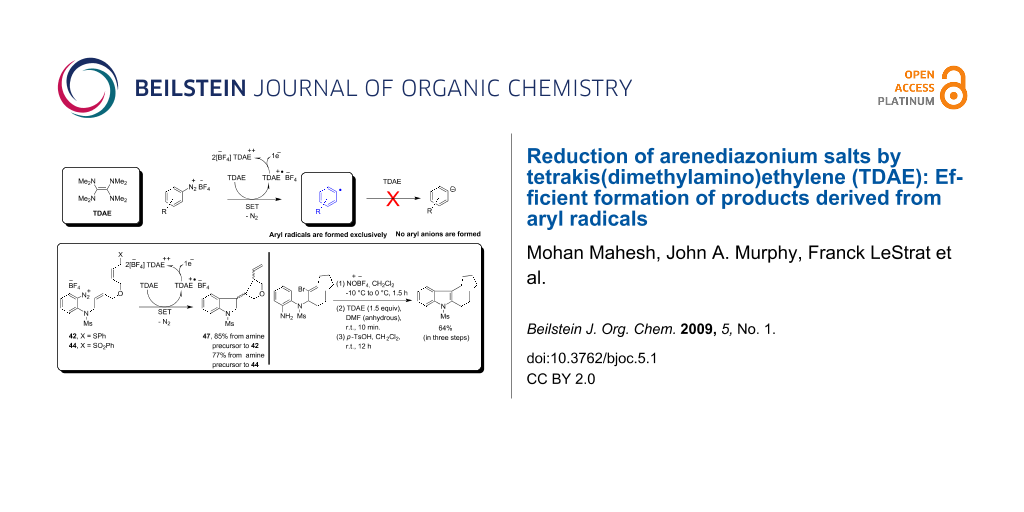
Graphical Abstract
Introduction
Arenediazonium salts have long proved useful as sources of aryl radicals in many reactions featuring carbon-carbon (e.g., Meerwein [1], Pschorr [2,3], Gomberg [3] reactions) and carbon-heteroatom bond (e.g., Sandmeyer [4]) formation. The radical-polar crossover reaction [5-15] of arenediazonium salts, developed in our group since 1993, also features aryl radical intermediates and is a more recent addition to these reactions. It involves a novel splicing of radical and polar reactions in one pot, employing tetrathiafulvalene (TTF, 4a, Scheme 1) as electron donor. A number of functionalised heterocycles [5-17] such as dihydrobenzofurans, indolines and indoles have been synthesized using this methodology and the radical-polar methodology has been employed successfully in the total synthesis [10] of aspidospermidine (15), the alkaloid of the Aspidosperma genus (Scheme 2). In line with our interests in generating aryl radicals by reduction of arenediazonium salts with tetrathiafulvalenes [5-16] 4 and dithiadiazafulvalenes [16,17] (DTDAF) 6 (Scheme 1) and later by electrochemical means [18], we were keen to compare the outcomes of these reactions of diazonium salts with those arising from the use of alternative neutral organic electron donors [19,20]. An interesting member of these alternative reagents is the commercially available and economically attractive tetrakis(dimethylamino)ethylene (TDAE, 1). This paper describes the results of our investigations on reactions of TDAE as a neutral organic electron donor with arenediazonium salts.
![[1860-5397-5-1-i1]](/bjoc/content/inline/1860-5397-5-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Aza- and thia-substituted electron donors.
Scheme 1: Aza- and thia-substituted electron donors.
TDAE (1), has been widely exploited as a strong electron donor [21-59] to electron-poor aliphatic and benzylic halides, notably those derived from organofluorine sources. Burkholder, Dolbier and Médebielle reported [28] that the electrochemical oxidation of TDAE in acetonitrile occurs in two reversible one-electron oxidation steps, to TDAE+• 2 and TDAE++ 3 at −0.78 V and −0.61 V vs saturated calomel electrode (SCE). Recently, TDAE-promoted reduction of electron-deficient o- and p-nitrobenzyl chlorides [44-47], 1,2-bis(bromomethyl)arenes [48], mono and trichloromethyl azaheterocycles [49,50], 2-(dibromomethyl)quinoxaline [51], α-bromoketones [52] have been reported. Vanelle and co-workers recently reported a photoinduced reduction of p-nitrobenzaldehyde in the presence of TDAE [53].
The utility of TDAE as a strong electron donor in specific organometallic reactions, such as the chromium-mediated allylation of aldehydes and ketones [54,55] and the palladium-catalyzed reductive homo-coupling of aryl halides to afford the corresponding biaryls [56-59] illustrate further versatility of the reagent.
The fact that formation of aryl radicals had never been reported using TDAE meant that we were keen to compare its reactions with those of the structurally related TTF (4a). Thus, as shown in Scheme 2, the radical-cation of TTF intercepts intermediates with the formation of C-S bonds in the radical-polar crossover reaction; would the analogous chemistry be seen with TDAE where no sulfur atoms are present?
![[1860-5397-5-1-i2]](/bjoc/content/inline/1860-5397-5-1-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical-polar crossover reaction of arenediazonium salts by TTF.
Scheme 2: Radical-polar crossover reaction of arenediazonium salts by TTF.
The experiments were of heightened interest because of the recent report by Andrieux and Pinson [60] on the standard reduction potential of the phenyl radical (formed by electrochemical reduction of the arenediazonium cation) to the phenyl anion (+0.05 V vs SCE). Thus, it had long been noted that cyclic voltammetry of aryl halides, particularly iodides, can give rise to a single two-electron wave in the reductive part of the cycle. The first electron converts the aryl iodide to the corresponding aryl radical, while the second electron transforms the aryl radical to an aryl anion. The two-electron single reductive wave arises because the transfer of the second electron is easier than the first. Andrieux and Pinson reasoned that in order to determine the potential for the conversion of aryl radical to aryl anion, a substrate other than an aryl halide would need to be used. As the one-electron reduction of an arenediazonium salt occurs [60] at much more positive potentials (Ep 0.16 V vs SCE) than for aryl iodides [61] (Ep −2.2 V vs SCE), this gives a much better chance to observe a second reductive peak in cyclic voltammetry and to determine the potential for conversion of aryl radicals to aryl anions. In the event, their study [60] showed two reductive peaks for benzenediazonium tetrafluoroborate (Ep 0.16 V and −0.64 V vs SCE). Through detailed analysis, Andrieux and Pinson showed that this second peak was consistent with reduction of the aryl radical to aryl anion and derived a value for the standard potential of this step as E0 = +0.05 V.
The reduction potentials determined by Andrieux and Pinson would be consistent with the chemistry that we had observed using TTF, in that TTF had been able to achieve the easier step of reducing arenediazonium salts to aryl radicals, but not the more difficult step (aryl radicals to aryl anions). The redox potentials associated with TTF are +0.32 V and +0.71 V vs SCE [62] so, even transferring one electron to the diazonium salts would superficially appear difficult, but it is well known that electron-transfer by a mediator in solution [63] is frequently more easily achieved (less negative potential) than would be expected from the bare electrochemical data. In the light of these facts, and given that TDAE is a much more powerful donor than TTF (by about 1.1 V for the transfer of the first electron), there is a danger that aryl radicals formed from arenediazonium salts using this reagent would be further converted into aryl anions, if the second electron transfer were sufficiently rapid. Therefore, we proposed to examine cyclization reactions of aryl radicals produced in this way to investigate this point.
Results and Discussion
Preparation of indolines
Our initial studies reacted TDAE with simple arenediazonium salt 16 (Scheme 3). On addition of TDAE to a solution of the arenediazonium tetrafluoroborate salt 16 in acetonitrile [Table 1, entries (i) and (ii)], or, alternately, on addition of the arenediazonium salt to excess TDAE (2.5 equiv) [Table 1, entry (iii)] the reaction mixture underwent effervescence as it turned from deep red to pale orange. In each case, the reaction yielded an inseparable mixture of indolines 20a and 20b in approximately equal yield. In entry (iii) of that table, these yields were estimated from a calibrated NMR determination; following this, the mixture was subjected to epoxidation with mCPBA, leading to isolation of 20b and the epoxide 20c (not shown in Scheme 3) derived from 20a. From this series of experiments, the oxidized product of TDAE, namely octamethyloxamidinium bis(tetrafluoroborate) (21) was isolated in up to 64% yield as an off-white powder. The structure of the salt 21 was characterized by NMR studies and also by mass spectrometry.
![[1860-5397-5-1-i3]](/bjoc/content/inline/1860-5397-5-1-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Studies on the reductive radical cyclization of arenediazonium salt 16 by TDAE.
Scheme 3: Studies on the reductive radical cyclization of arenediazonium salt 16 by TDAE.
Table 1: Reductive radical cyclization of arenediazonium salt 16 by TDAE.
| Entry | Equivalents of TDAE | Solvent | Temperature (°C) | Time | Yield (%) of | ||
|---|---|---|---|---|---|---|---|
| 20a | 20b | 21 | |||||
| (i) | 1.0 | Acetonitrile | 25 | 24 h | 11 | 11 | 50 |
| (ii) | 1.0 | Methanol | −40 to 25 | 24 h | 4 | 5 | 64 |
| (iii) | 2.5 | Acetonitrile | 25 | 10 min | 12 | 17 | –a |
a[TDAE]++ 2[BF4−] salt 21 was not isolated from the reaction
The formation of the indolines 20a and 20b could then be envisaged through an intermolecular radical disproportionation reaction of two cyclised radical intermediates 19 as explained in Scheme 3. No evidence was seen for the formation of salt 22 although the yields of the products 20 were not high. Non-observation of 22 illustrates that TDAE+•, unlike TTF+•, does not provide an efficient termination of radical processes, and the low yields of isolated compounds could be consistent with radical chemistry where efficient termination was lacking. With this as guidance to our thinking, the remaining substrates below were designed to provide internal termination routes for the radical chemistry.
One way to achieve clean termination of the radical process would be by providing a radical leaving group adjacent to the cyclised radical 19. Appropriate groups might be sulfide, sulfoxide and sulfonyl groups [64,65]. Accordingly, arenediazonium salts 31a–d were prepared bearing appropriate terminal radical leaving groups (Scheme 4) and treated with 1 equivalent of TDAE under different solvent conditions and temperature. As expected, the aryl radical generated from the reduction of the arenediazonium salts, underwent facile self-terminating 5-exo-trig aryl radical-alkene cyclization to afford the indolines 20a, 32 as the sole products in very high yields (Table 2). Owing to the sensitive nature of the arenediazonium salts 31a–d, they were usually generated in situ from the corresponding amines 30a–d by treatment with nitrosonium tetrafluoroborate. One of the notable features of these cyclizations is the ease of purification of the product from the reaction mixture. The oxidized product of TDAE, namely octamethyloxamidinium bis(tetrafluoroborate) (21), was easily removed either by filtration or by simple work-up with water.
![[1860-5397-5-1-i4]](/bjoc/content/inline/1860-5397-5-1-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of the arenediazonium salts 31a–d. Reagents and conditions: (a) 23, NaH, THF, 0 °C, 0.5 h, then TBDPS-Cl, 0 °C to 25 °C, 4 h, 85%; (b) 25, DIAD, PPh3, THF, 0 °C to 25 °C, 12 h, 80% (26a), 64% (26d); (c) TBAF, THF, 25 °C, 1.5 h, 85% (27a), 0.5 h, 97% (27d); (d) PBr3, DCM, 0 °C to 25 °C, 1 h, 99% (28a), 93% (28d); (e) PhSH, NaH, THF, 0 °C to 25 °C, 1 h, then 28, 25 °C, 5 h, 98% (29a), 94% (29d); (f) Cu(acac)2, NaBH4, EtOH, 25 °C, 15 h, 70% (30a), SnCl2·2H2O, MeOH, 65 °C, 4 h, 91% (30d); (g) NOBF4, CH2Cl2, −20 °C to −10 °C, 1.5 h, 98% (31a), 100% (31d); (h) NaIO4, MeOH/H2O, r.t., 1 h 15 min, 92%; (i) SnCl2, MeOH, 65 °C, 4 h, 71%; (j) NOBF4, CH2Cl2, −20 °C to −10 °C, 1.5 h, 100%; (k) NaIO4, MeOH/H2O (1:1), r.t., 72 h, 82%; (l) SnCl2·2H2O, MeOH, 3.5 h, 83%; (m) NOBF4, CH2Cl2, −20 °C to −10 °C, 1.5 h, 100%.
Scheme 4: Preparation of the arenediazonium salts 31a–d. Reagents and conditions: (a) 23, NaH, THF, 0 °C, 0.5...
Table 2: Reductive radical cyclization of arenediazonium salts 31a–d by TDAE.
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-1-i11.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Diazonium Salt | Equivalents of TDAE | Solvent | Temperature (°C) | Time | Isolated Yield (%) of | ||
|---|---|---|---|---|---|---|---|---|
| 20a/32 | 33 | 21 | ||||||
| (i) | 31a | 1.0 | Acetonitrile | 25 | 5 min | 88 | 82 | 12 |
| (ii)a | 31a | 0 | Acetonitrile | 25 | 5 min | 0 | 0 | 0 |
| (iii)b,d | 31a | 3.0 | Acetonitrile | 25 | 5 min | 85 | 39 | –c |
| (iv) | 31a | 1.0 | Acetone | 25 | 5 min | 84 | 78 | 17 |
| (v) | 31a | 1.0 | Methanol | 0 to 25 | 2 h | 81 | 34 | 71 |
| (vi)d | 31b | 1.0 | Acetone | 25 | 5 min | 81 | 0 | 24 |
| (vii)d | 31c | 1.0 | Acetone | 25 | 5 min | 63 | 0 | 41 |
| (viii)d | 31d | 1.0 | Acetone | 25 | 5 min | 88 | 55 | 35 |
aControl reaction performed in the absence of TDAE reagent. bThis experiment was conducted by adding a solution of the diazonium salt in dry MeCN to a solution of TDAE in dry MeCN, while the other experiments in this table all featured addition of the TDAE to the diazonium salt. c[TDAE]++ 2[BF4−] salt 21 was not isolated from the reaction. dIn these experiments, the arenediazonium salts 31a–d were made in situ from their corresponding amines 30a–d, while all the other experiments in this table featured direct use of arenediazonium salt.
Cascade cyclizations
To determine the scope of the TDAE-mediated reduction of arenediazonium salts, we sought to extend this methodology to more complex substrates, namely 42 and 44. Pleasingly, the arenediazonium salts 42 and 44, prepared in situ from the amines 41 and 43 respectively upon treatment with 1 equivalent of TDAE, underwent facile cascade radical cyclizations to afford the bicyclized product 47 in 85% and 77% yield respectively (Scheme 5). The ability of TDAE to mediate such efficient cascade cyclizations via two C-C bond formations reactions in one pot from the aryl radical 45 was significant considering the fact that our previous studies on similar substrates by TTF [5,8,16,17] and TMTTF [16] had shown competitive trapping of the intermediate alkyl radical 46 by TTF+• or TMTTF+•.
![[1860-5397-5-1-i5]](/bjoc/content/inline/1860-5397-5-1-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cascade radical cyclizations of arenediazonium salts 42 and 44 by TDAE. Reagents and conditions: (a) 23, NaH, THF, 0 °C, 0.5 h, then TBDPS-Cl, 0 °C to 25 °C, 4 h, 85%; (b) NBS, Me2S, CH2Cl2, −30 °C, 0.5 h, then 24, −30 °C to r.t., 3 h, 95%; (c) 23, NaH, THF, 0 °C to r.t., 1 h, then 34, 72 h, 61%; (d) N-(2-nitrophenyl)methanesulfonamide (25a), DIAD, PPh3, THF, 0 °C to r.t., 1 h, 98%; (e) TBAF, THF, r.t., 20 min, 90%; (f) NBS, PPh3, CH2Cl2, −25 °C, 20 min, then 37, −25 °C to r.t., 40 min, 96%; (g) PhSH, NaH, THF, 0 °C to r.t., 1 h, then 38, r.t., 12 h, 86%; (h) NaIO4, H2O, MeOH, r.t., 76 h, 73%; (i) SnCl2, CH3OH, H2O, reflux, 3.5 h to 4 h, 95% (41), 78% (43); (j) NOBF4, CH2Cl2, −15 °C to 0 °C, 1.5 h; (k) NOBF4, CH2Cl2, −15 °C to 0 °C, 1.5 h; (l) TDAE (1.0 equiv), acetone, 10 min, r.t., 85% 47, 46% 33, 52% 21 in two steps, from 41; 77% 47, 46% 21 in two steps, from 43.
Scheme 5: Cascade radical cyclizations of arenediazonium salts 42 and 44 by TDAE. Reagents and conditions: (a...
Preparation of indoles
Following the successful implementation of the methodology on the synthesis of indolines, we next sought to harness the aryl radicals in the synthesis of indoles by a radical-based addition-elimination strategy [66,67]. However, our initial attempts in this area upon cyclization of arenediazonium salt 49a were not fruitful as the reactions afforded a mixture of the exocyclic alkene 50a and the alcohol 52a [Table 3, entry (i)]. We expected that the aryl radical 53a generated by the reduction of arenediazonium salt 49a by TDAE would undergo 5-exo-trig radical cyclization onto the vinyl bromide to afford the alkyl radical intermediate 54a, from which Br• would be eliminated to afford the exocyclic alkene 50a (Scheme 6). Such alkenes tautomerise easily to the corresponding indoles (in this case 51a) in the presence of a trace of acid. Alcohol 52a can arise by 1,2-bromine shift [66,68,69] from radical 54a followed by either (a) loss a hydrogen atom from the resulting benzylic radical 56a in collision with another radical or, less likely, (b) oxidation of 56a by electron transfer to an arenediazonium cation. Loss of a proton from the cation so formed would yield bromoalkylindole 57a and subsequent hydrolysis would result in the alcohol 52a. However, when the same reaction was re-examined in anhydrous DMF as the solvent with 1.5 equivalents of TDAE, it afforded the unstable exocyclic alkene 50a as the sole product, which after treatment with p-toluenesulfonic acid tautomerised to the indole 51a in an overall 68% yield in three steps from 48a. Adopting the optimized procedure, the diazonium salts 49b–d on treatment with 1.5 equivalents of TDAE in anhydrous DMF yielded the indoles 51b–d in 63%, 43% and 64% yields (in three steps from the corresponding aryl amines 48b–d) respectively (Scheme 7). The indole 51d bearing a fused 9-membered ring was of particular interest to us because the important anticancer drugs vinblastine (58a) and vincristine (58b) contain such a system.
Table 3: Initial optimization studies of cyclization of arenediazonium salt 49a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-1-i12.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Diazonium Salt | Equivalents of TDAE | Solvent | Temperature (°C) | Time | Isolated Yielda (%) of | |
|---|---|---|---|---|---|---|---|
| 51a | 52a | ||||||
| (i) | 49a | 1.0 | Acetone | 25 | 30 min | 39b | 40 |
| (ii) | 49a | 1.5 | DMF (anhydrous) | 25 | 10 min | 68c | 0 |
aAll isolated yields were calculated on the basis of the quantity of the starting aryl amine 48a. bThe product 51a was isolated directly from the reaction mixture after auto-tautomerisation of 50a to indole 51a during storage of reaction mixture prior to flash chromatography. cThe product 51a was obtained by treatment of the intermediate exocyclic alkene 50a with p-toluenesulfonic acid monohydrate in dichloromethane at r.t. for 12 h.
![[1860-5397-5-1-i6]](/bjoc/content/inline/1860-5397-5-1-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: TDAE-mediated radical based addition-elimination route to indoles.
Scheme 6: TDAE-mediated radical based addition-elimination route to indoles.
![[1860-5397-5-1-i7]](/bjoc/content/inline/1860-5397-5-1-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Cyclization of the arenediazonium salts 49b–d by TDAE. Reagents and conditions: (a) NOBF4, CH2Cl2, −10 °C to 0 °C, 1.5 h; (b) TDAE (1.5 equiv), anhydrous DMF, r.t., 10 min; (c) p-toluenesulfonic acid monohydrate, CH2Cl2, r.t., 12 h, 63% (51b, in three steps from 48b), 43% (51c, in three steps from 48c); (d) NOBF4, CH2Cl2, −10 °C to 0 °C, 1.5 h; (e) TDAE (1.5 equiv), anhydrous DMF, r.t., 10 min, 74% 50d; (f) p-toluenesulfonic acid monohydrate, CH2Cl2, r.t., 12 h, 64% (overall yield in three steps, from 48d).
Scheme 7: Cyclization of the arenediazonium salts 49b–d by TDAE. Reagents and conditions: (a) NOBF4, CH2Cl2, ...
Aryl C-C bond formation
As a final extension of this methodology, we probed the feasibility of this methodology in aryl-aryl C-C bond formation reactions. Accordingly, the diazonium salt 62 was prepared from indoline (59), and treated with one equivalent of TDAE in acetone as solvent. The reaction mixture instantaneously turned deep red, with accompanying effervescence of nitrogen, and afforded the tetracyclic sulfonamide 65 in 60% yield along with indole (63) and indole sulfonamide 64 in 33% and 5% yield respectively (Scheme 8).
![[1860-5397-5-1-i8]](/bjoc/content/inline/1860-5397-5-1-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Cyclization of the arenediazonium salt 62 by TDAE. Reagents and conditions: (a) 2-Nitrobenzenesulfonyl chloride, DMAP, pyridine, 0 °C then 110 °C, 27 h, 57%; (b) H2, Pd/C, EtOAc, 3 h, 98%; (c) 61, NOBF4, CH2Cl2, −10 °C, 1.5 h, 95%; (d) TDAE, CH3OH/acetone (1:1), 25 °C, 10 min, 33% 63, 5% 64, 60% 65.
Scheme 8: Cyclization of the arenediazonium salt 62 by TDAE. Reagents and conditions: (a) 2-Nitrobenzenesulfo...
Initial SET from TDAE to the arenediazonium salt 62 afforded the aryl radical 67, with release of molecular nitrogen. The aryl radical 67 would be expected [3,11,70] to cyclise onto the aryl ring A either through 5-exo or 6-endo cyclization. The radicals 68 and 69 could interconvert. Alternatively, aryl radical 67 could also undergo direct formation of the radical 69. Rearomatization from 69 might then occur through a number of pathways; in one of these, the radical intermediate 69 would lose a proton to yield the radical anion 70 which, upon oxidation by loss of single electron to the starting diazonium salt 62, would result in the formation of the tetracyclic sulfonamide 65 (Scheme 9).
![[1860-5397-5-1-i9]](/bjoc/content/inline/1860-5397-5-1-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Mechanism for the formation of the tetracyclic sulfonamide 65.
Scheme 9: Mechanism for the formation of the tetracyclic sulfonamide 65.
Indole (63) and indole sulfonamide 64 can be formed via the indolinyl radical intermediate 71 (Scheme 10). The formation of the indolinyl radical 71 could be envisaged by abstraction of the hydrogen atom (1,5-hydrogen translocation) by the aryl radical 67 from the carbon atom in the α-position to the nitrogen atom of the indoline nucleus within the same molecule. The indolinyl radical 71 might follow pathway A and undergo radical fragmentation to the intermediate 75 which would eventually tautomerise to indole (63). The precedent for this radical fragmentation of the sulfonyl group comes from the previous work of our group [71], where a similar indolinyl radical underwent a radical cleavage of N-S bond to eliminate the sulfonyl group.
![[1860-5397-5-1-i10]](/bjoc/content/inline/1860-5397-5-1-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Possible mechanism for the formation of indole (63) and indole sulfonamide 64.
Scheme 10: Possible mechanism for the formation of indole (63) and indole sulfonamide 64.
Indole sulfonamide 64 could be explained by deprotonation of radical 71 by TDAE to form radical-anion 72, followed by electron loss. Alternatively, removal of hydrogen atom through reaction with another radical could afford 64.
Conclusion
We have reported a mild and direct method for generation of aryl radicals by reduction of arenediazonium salts using TDAE as a neutral ground-state organic electron donor. Additionally, we have described the utility of the aryl radicals in the construction of indolines and indoles by intramolecular radical cyclization of aryl radicals onto appropriately placed alkenes bearing terminal radical leaving groups. The presence of a suitable radical leaving group like a sulfide, sulfoxide or sulfone is necessary for the self-termination of the 5-exo-trig radical cyclization reactions to avoid competing intermolecular radical side-reactions. The TDAE-mediated radical-based addition-elimination route for the construction of indole ring systems warranted anhydrous reaction conditions for greater efficiency. A preliminary study on TDAE-mediated aryl-aryl C-C bond formation reaction has also been discussed. TDAE possesses a distinct advantage over other organic reducing agents as the oxidized products of TDAE are water soluble – thus the purification process is highly convenient. Further extensions of this methodology in the construction of several heterocyclic ring systems and complex synthetic targets for natural product synthesis are currently in progress in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental data | ||
| Format: DOC | Size: 1.8 MB | Download |
Acknowledgments
We thank Universities UK (CVCP) for the award of Overseas Research Scholarship (ORS), University of Strathclyde for an International Research Scholarship, and F. Hoffmann-La Roche, Ltd., Basel for a CASE award to M.M.; EPSRC for funding; EPSRC National Mass Spectrometry Service, Swansea for the mass spectral analysis and Dr. John A. Parkinson, University of Strathclyde for NOE NMR experiments.
References
-
Meerwein, H.; Buchner, E.; van Emster, K. J. Prakt. Chem. 1939, 152, 237. doi:10.1002/prac.19391520705
Return to citation in text: [1] -
Abramovitch, R. A. Adv. Free-Radical Chem. 1966, 2, 87.
Return to citation in text: [1] -
Sainsbury, M. Tetrahedron 1980, 36, 3327. doi:10.1016/0040-4020(80)80185-2
Return to citation in text: [1] [2] [3] -
Hanson, P.; Hammond, R. C.; Goodacre, P. R.; Purcell, J.; Timms, A. W. J. Chem. Soc., Perkin Trans. 2 1994, 691. doi:10.1039/p29940000691
Return to citation in text: [1] -
Bashir, N.; Patro, B.; Murphy, J. A. In Advances in Free Radical Chemistry, Vol. 2; Zard, S. Z., Ed.; JAI Press: Stamford, CT, U.S.A., 1999; pp 123–150.
Return to citation in text: [1] [2] [3] [4] -
Murphy, J. A. The Radical-Polar Crossover Reaction. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 298–315. doi:10.1002/9783527618293.ch15
Return to citation in text: [1] [2] [3] -
Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295. doi:10.1039/C39930000295
Return to citation in text: [1] [2] [3] -
Fletcher, R. J.; Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1995, 623. doi:10.1039/P19950000623
Return to citation in text: [1] [2] [3] [4] -
Murphy, J. A.; Rasheed, F.; Gastaldi, S.; Ravishanker, T.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1997, 1549. doi:10.1039/a607060d
Return to citation in text: [1] [2] [3] -
Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995. doi:10.1039/a900335e
Return to citation in text: [1] [2] [3] [4] -
Lampard, C.; Murphy, J. A.; Rasheed, F.; Lewis, N.; Hursthouse, M. B.; Hibbs, D. E. Tetrahedron Lett. 1994, 35, 8675. doi:10.1016/S0040-4039(00)78469-3
Return to citation in text: [1] [2] [3] [4] -
Murphy, J. A.; Rasheed, F.; Roome, S. J.; Lewis, N. Chem. Commun. 1996, 737. doi:10.1039/cc9960000737
Return to citation in text: [1] [2] [3] -
Fletcher, R.; Kizil, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. J. Chem. Soc., Perkin Trans. 1 1998, 2341. doi:10.1039/a802974a
Return to citation in text: [1] [2] [3] -
Kizil, M.; Lampard, C.; Murphy, J. A. Tetrahedron Lett. 1996, 37, 2511. doi:10.1016/0040-4039(96)00306-1
Return to citation in text: [1] [2] [3] -
Fletcher, R. J.; Hibbs, D. E.; Hursthouse, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. Chem. Commun. 1996, 739. doi:10.1039/cc9960000739
Return to citation in text: [1] [2] [3] -
Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3
Return to citation in text: [1] [2] [3] [4] [5] -
Koizumi, T.; Bashir, N.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 3637. doi:10.1039/a906684e
Return to citation in text: [1] [2] [3] -
LeStrat, F.; Murphy, J. A.; Hughes, M. Org. Lett. 2002, 4, 2735. doi:10.1021/ol0262643
Return to citation in text: [1] -
Murphy, J. A.; Khan, T. A.; Zhou, S.-z.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356. doi:10.1002/anie.200462038
Return to citation in text: [1] -
Murphy, J. A.; Zhou, S.-z.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178. doi:10.1002/anie.200700554
Return to citation in text: [1] -
Wiberg, N. Angew. Chem., Int. Ed. Engl. 1968, 7, 766. doi:10.1002/anie.196807661
Return to citation in text: [1] -
Fritsch, J. M.; Weingarten, H.; Wilson, J. D. J. Am. Chem. Soc. 1970, 92, 4038. doi:10.1021/ja00716a035
Return to citation in text: [1] -
Kolomeitsev, A.; Médebielle, M.; Kirsch, P.; Lork, E.; Röschenthaler, G.-V. J. Chem. Soc., Perkin Trans. 1 2000, 2183. doi:10.1039/b002252g
Return to citation in text: [1] -
Carpenter, W.; Bens, E. M. Tetrahedron 1970, 26, 59. doi:10.1016/0040-4020(70)85007-4
Return to citation in text: [1] -
Winberg, H. E.; Downing, J. R.; Coffman, D. D. J. Am. Chem. Soc. 1965, 87, 2054. doi:10.1021/ja01087a039
Return to citation in text: [1] -
Wiberg, N.; Buchler, J. W. Angew. Chem., Int. Ed. Engl. 1962, 1, 406. doi:10.1002/anie.196204061
Return to citation in text: [1] -
Wiberg, N.; Buchler, J. W. Chem. Ber. 1963, 96, 3223. doi:10.1002/cber.19630961218
Return to citation in text: [1] -
Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 1998, 63, 5385. doi:10.1021/jo980201+
Return to citation in text: [1] [2] -
Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M.; Ndedi, A. Tetrahedron Lett. 1998, 39, 8853. doi:10.1016/S0040-4039(98)02028-0
Return to citation in text: [1] -
Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M.; Aït-Mohand, S. Tetrahedron Lett. 2001, 42, 3077. doi:10.1016/S0040-4039(01)00388-4
Return to citation in text: [1] -
Médebielle, M.; Keirouz, R.; Okada, E.; Ashida, T. Synlett 2001, 821. doi:10.1055/s-2001-14594
Return to citation in text: [1] -
Aït-Mohand, S.; Takechi, N.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2001, 3, 4271. doi:10.1021/ol016933x
Return to citation in text: [1] -
Takechi, N.; Aït-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Tetrahedron Lett. 2002, 43, 4317. doi:10.1016/S0040-4039(02)00800-6
Return to citation in text: [1] -
Takechi, N.; Aït-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 4671. doi:10.1021/ol0270374
Return to citation in text: [1] -
Médebielle, M.; Kato, K.; Dolbier, W. R., Jr. Tetrahedron Lett. 2003, 44, 7871. doi:10.1016/j.tetlet.2003.09.020
Return to citation in text: [1] -
Médebielle, M.; Hohn, S.; Okada, E.; Myoken, H.; Shibata, D. Tetrahedron Lett. 2005, 46, 7817. doi:10.1016/j.tetlet.2005.09.018
Return to citation in text: [1] -
Prakash, G. K. S.; Wang, Y.; Hu, J.; Olah, G. A. J. Fluorine Chem. 2005, 126, 1361. doi:10.1016/j.jfluchem.2005.07.011
Return to citation in text: [1] -
Xu, W.; Dolbier, W. R., Jr. J. Org. Chem. 2005, 70, 4741. doi:10.1021/jo050483v
Return to citation in text: [1] -
Peng, W.; Zhao, J.; He, P.; Zhu, S. Synlett 2006, 296. doi:10.1055/s-2006-926225
Return to citation in text: [1] -
Peng, W.; Zhao, J.; Zhu, S. J. Fluorine Chem. 2006, 127, 360. doi:10.1016/j.jfluchem.2005.12.017
Return to citation in text: [1] -
Peng, W.; Zhao, J.; Zhu, S. Synthesis 2006, 1470. doi:10.1055/s-2006-926436
Return to citation in text: [1] -
Pooput, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 2006, 71, 3564. doi:10.1021/jo060250j
Return to citation in text: [1] -
Médebielle, M.; Keirouz, R.; Okada, E.; Shibata, D.; Dolbier, W. R., Jr. Tetrahedron Lett. 2008, 49, 589. doi:10.1016/j.tetlet.2007.11.146
Return to citation in text: [1] -
Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2003, 44, 6433. doi:10.1016/S0040-4039(03)01594-6
Return to citation in text: [1] [2] -
Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2004, 45, 5121. doi:10.1016/j.tetlet.2004.04.166
Return to citation in text: [1] [2] -
Giuglio-Tonolo, G.; Terme, T.; Vanelle, P. Synlett 2005, 251. doi:10.1055/s-2004-837227
Return to citation in text: [1] [2] -
Amiri-Attou, O.; Terme, T.; Vanelle, P. Synlett 2005, 3047. doi:10.1055/s-2005-921916
Return to citation in text: [1] [2] -
Nishiyama, Y.; Kawabata, H.; Kobayashi, A.; Nishino, T.; Sonoda, N. Tetrahedron Lett. 2005, 46, 867. doi:10.1016/j.tetlet.2004.11.114
Return to citation in text: [1] [2] -
Montana, M.; Crozet, M. D.; Castera-Ducros, C.; Terme, T.; Vanelle, P. Heterocycles 2008, 75, 925.
Return to citation in text: [1] [2] -
Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2006, 47, 6573. doi:10.1016/j.tetlet.2006.07.030
Return to citation in text: [1] [2] -
Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2005, 46, 8373. doi:10.1016/j.tetlet.2005.09.152
Return to citation in text: [1] [2] -
Nishiyama, Y.; Kobayashi, A. Tetrahedron Lett. 2006, 47, 5565. doi:10.1016/j.tetlet.2006.05.141
Return to citation in text: [1] [2] -
Amiri-Attou, O.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2008, 49, 1016. doi:10.1016/j.tetlet.2007.12.011
Return to citation in text: [1] [2] -
Kuroboshi, M.; Goto, K.; Mochizuki, M.; Tanaka, H. Synlett 1999, 1930. doi:10.1055/s-1999-2995
Return to citation in text: [1] [2] -
Kuroboshi, M.; Tanaka, M.; Kishimoto, S.; Goto, K.; Mochizuki, M.; Tanaka, H. Tetrahedron Lett. 2000, 41, 81. doi:10.1016/S0040-4039(99)02006-7
Return to citation in text: [1] [2] -
Kuroboshi, M.; Waki, Y.; Tanaka, H. Synlett 2002, 637. doi:10.1055/s-2002-22719
Return to citation in text: [1] [2] -
Kuroboshi, M.; Waki, Y.; Tanaka, H. J. Org. Chem. 2003, 68, 3938. doi:10.1021/jo0207473
Return to citation in text: [1] [2] -
Park, S. B.; Alper, H. Tetrahedron Lett. 2004, 45, 5515. doi:10.1016/j.tetlet.2004.05.013
Return to citation in text: [1] [2] -
Kuroboshi, M.; Takeda, T.; Motoki, R.; Tanaka, H. Chem. Lett. 2005, 34, 530. doi:10.1246/cl.2005.530
Return to citation in text: [1] [2] -
Andrieux, C. P.; Pinson, J. J. Am. Chem. Soc. 2003, 125, 14801. doi:10.1021/ja0374574
Return to citation in text: [1] [2] [3] -
Pause, L.; Robert, M.; Savéant, J.-M. J. Am. Chem. Soc. 1999, 121, 7158. doi:10.1021/ja991365q
Return to citation in text: [1] -
Ashton, P. R.; Balzani, V.; Becher, J.; Credi, A.; Fyfe, M. C. T.; Mattersteig, G.; Menzer, S.; Nielsen, M. B.; Raymo, F. M.; Stoddart, J. F.; Venturi, M.; Willliams, D. J. J. Am. Chem. Soc. 1999, 121, 3951. doi:10.1021/ja984341c
Return to citation in text: [1] -
Simonet, J.; Pilard, J.-F. Electrogenerated Reagents. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker Inc.: New York, 1991; pp 1163–1225.
See in particular pp 1171 ff.
Return to citation in text: [1] -
Lacôte, E.; Delouvrié, B.; Fensterbank, L.; Malacria, M. Angew. Chem., Int. Ed. 1998, 37, 2116. doi:10.1002/(SICI)1521-3773(19980817)37:15<2116::AID-ANIE2116>3.0.CO;2-L
Return to citation in text: [1] -
Wagner, P. J.; Sedon, J. H.; Lindstrom, M. J. J. Am. Chem. Soc. 1978, 100, 2579. doi:10.1021/ja00476a068
Return to citation in text: [1] -
Murphy, J. A.; Scott, K. A.; Sinclair, R. S.; Martin, C. G.; Kennedy, A. R.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 2000, 2395. doi:10.1039/b002565h
Return to citation in text: [1] [2] -
Murphy, J. A.; Scott, K. A.; Sinclair, R. S.; Lewis, N. Tetrahedron Lett. 1997, 38, 7295. doi:10.1016/S0040-4039(97)01695-X
Return to citation in text: [1] -
Freidlina, R. Kh.; Terent’ev, A. B. Adv. Free-Radical Chem. 1980, 6, 1.
Return to citation in text: [1] -
Freidlina, R. Kh. Adv. Free-Radical Chem. 1965, 1, 211.
Return to citation in text: [1] -
Bowman, W. R.; Heaney, H.; Jordan, B. M. Tetrahedron 1991, 47, 10119. doi:10.1016/S0040-4020(01)96061-2
Return to citation in text: [1] -
Bommezijn, S.; Martin, C. G.; Kennedy, A. R.; Lizos, D.; Murphy, J. A. Org. Lett. 2001, 3, 3405. doi:10.1021/ol0166449
Return to citation in text: [1]
| 64. | Lacôte, E.; Delouvrié, B.; Fensterbank, L.; Malacria, M. Angew. Chem., Int. Ed. 1998, 37, 2116. doi:10.1002/(SICI)1521-3773(19980817)37:15<2116::AID-ANIE2116>3.0.CO;2-L |
| 65. | Wagner, P. J.; Sedon, J. H.; Lindstrom, M. J. J. Am. Chem. Soc. 1978, 100, 2579. doi:10.1021/ja00476a068 |
| 5. | Bashir, N.; Patro, B.; Murphy, J. A. In Advances in Free Radical Chemistry, Vol. 2; Zard, S. Z., Ed.; JAI Press: Stamford, CT, U.S.A., 1999; pp 123–150. |
| 8. | Fletcher, R. J.; Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1995, 623. doi:10.1039/P19950000623 |
| 16. | Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3 |
| 17. | Koizumi, T.; Bashir, N.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 3637. doi:10.1039/a906684e |
| 16. | Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3 |
| 1. | Meerwein, H.; Buchner, E.; van Emster, K. J. Prakt. Chem. 1939, 152, 237. doi:10.1002/prac.19391520705 |
| 5. | Bashir, N.; Patro, B.; Murphy, J. A. In Advances in Free Radical Chemistry, Vol. 2; Zard, S. Z., Ed.; JAI Press: Stamford, CT, U.S.A., 1999; pp 123–150. |
| 6. | Murphy, J. A. The Radical-Polar Crossover Reaction. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 298–315. doi:10.1002/9783527618293.ch15 |
| 7. | Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295. doi:10.1039/C39930000295 |
| 8. | Fletcher, R. J.; Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1995, 623. doi:10.1039/P19950000623 |
| 9. | Murphy, J. A.; Rasheed, F.; Gastaldi, S.; Ravishanker, T.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1997, 1549. doi:10.1039/a607060d |
| 10. | Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995. doi:10.1039/a900335e |
| 11. | Lampard, C.; Murphy, J. A.; Rasheed, F.; Lewis, N.; Hursthouse, M. B.; Hibbs, D. E. Tetrahedron Lett. 1994, 35, 8675. doi:10.1016/S0040-4039(00)78469-3 |
| 12. | Murphy, J. A.; Rasheed, F.; Roome, S. J.; Lewis, N. Chem. Commun. 1996, 737. doi:10.1039/cc9960000737 |
| 13. | Fletcher, R.; Kizil, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. J. Chem. Soc., Perkin Trans. 1 1998, 2341. doi:10.1039/a802974a |
| 14. | Kizil, M.; Lampard, C.; Murphy, J. A. Tetrahedron Lett. 1996, 37, 2511. doi:10.1016/0040-4039(96)00306-1 |
| 15. | Fletcher, R. J.; Hibbs, D. E.; Hursthouse, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. Chem. Commun. 1996, 739. doi:10.1039/cc9960000739 |
| 48. | Nishiyama, Y.; Kawabata, H.; Kobayashi, A.; Nishino, T.; Sonoda, N. Tetrahedron Lett. 2005, 46, 867. doi:10.1016/j.tetlet.2004.11.114 |
| 4. | Hanson, P.; Hammond, R. C.; Goodacre, P. R.; Purcell, J.; Timms, A. W. J. Chem. Soc., Perkin Trans. 2 1994, 691. doi:10.1039/p29940000691 |
| 49. | Montana, M.; Crozet, M. D.; Castera-Ducros, C.; Terme, T.; Vanelle, P. Heterocycles 2008, 75, 925. |
| 50. | Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2006, 47, 6573. doi:10.1016/j.tetlet.2006.07.030 |
| 28. | Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 1998, 63, 5385. doi:10.1021/jo980201+ |
| 2. | Abramovitch, R. A. Adv. Free-Radical Chem. 1966, 2, 87. |
| 3. | Sainsbury, M. Tetrahedron 1980, 36, 3327. doi:10.1016/0040-4020(80)80185-2 |
| 44. | Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2003, 44, 6433. doi:10.1016/S0040-4039(03)01594-6 |
| 45. | Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2004, 45, 5121. doi:10.1016/j.tetlet.2004.04.166 |
| 46. | Giuglio-Tonolo, G.; Terme, T.; Vanelle, P. Synlett 2005, 251. doi:10.1055/s-2004-837227 |
| 47. | Amiri-Attou, O.; Terme, T.; Vanelle, P. Synlett 2005, 3047. doi:10.1055/s-2005-921916 |
| 16. | Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3 |
| 17. | Koizumi, T.; Bashir, N.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 3637. doi:10.1039/a906684e |
| 19. | Murphy, J. A.; Khan, T. A.; Zhou, S.-z.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356. doi:10.1002/anie.200462038 |
| 20. | Murphy, J. A.; Zhou, S.-z.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178. doi:10.1002/anie.200700554 |
| 3. | Sainsbury, M. Tetrahedron 1980, 36, 3327. doi:10.1016/0040-4020(80)80185-2 |
| 11. | Lampard, C.; Murphy, J. A.; Rasheed, F.; Lewis, N.; Hursthouse, M. B.; Hibbs, D. E. Tetrahedron Lett. 1994, 35, 8675. doi:10.1016/S0040-4039(00)78469-3 |
| 70. | Bowman, W. R.; Heaney, H.; Jordan, B. M. Tetrahedron 1991, 47, 10119. doi:10.1016/S0040-4020(01)96061-2 |
| 5. | Bashir, N.; Patro, B.; Murphy, J. A. In Advances in Free Radical Chemistry, Vol. 2; Zard, S. Z., Ed.; JAI Press: Stamford, CT, U.S.A., 1999; pp 123–150. |
| 6. | Murphy, J. A. The Radical-Polar Crossover Reaction. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 298–315. doi:10.1002/9783527618293.ch15 |
| 7. | Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295. doi:10.1039/C39930000295 |
| 8. | Fletcher, R. J.; Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1995, 623. doi:10.1039/P19950000623 |
| 9. | Murphy, J. A.; Rasheed, F.; Gastaldi, S.; Ravishanker, T.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1997, 1549. doi:10.1039/a607060d |
| 10. | Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995. doi:10.1039/a900335e |
| 11. | Lampard, C.; Murphy, J. A.; Rasheed, F.; Lewis, N.; Hursthouse, M. B.; Hibbs, D. E. Tetrahedron Lett. 1994, 35, 8675. doi:10.1016/S0040-4039(00)78469-3 |
| 12. | Murphy, J. A.; Rasheed, F.; Roome, S. J.; Lewis, N. Chem. Commun. 1996, 737. doi:10.1039/cc9960000737 |
| 13. | Fletcher, R.; Kizil, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. J. Chem. Soc., Perkin Trans. 1 1998, 2341. doi:10.1039/a802974a |
| 14. | Kizil, M.; Lampard, C.; Murphy, J. A. Tetrahedron Lett. 1996, 37, 2511. doi:10.1016/0040-4039(96)00306-1 |
| 15. | Fletcher, R. J.; Hibbs, D. E.; Hursthouse, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. Chem. Commun. 1996, 739. doi:10.1039/cc9960000739 |
| 16. | Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3 |
| 21. | Wiberg, N. Angew. Chem., Int. Ed. Engl. 1968, 7, 766. doi:10.1002/anie.196807661 |
| 22. | Fritsch, J. M.; Weingarten, H.; Wilson, J. D. J. Am. Chem. Soc. 1970, 92, 4038. doi:10.1021/ja00716a035 |
| 23. | Kolomeitsev, A.; Médebielle, M.; Kirsch, P.; Lork, E.; Röschenthaler, G.-V. J. Chem. Soc., Perkin Trans. 1 2000, 2183. doi:10.1039/b002252g |
| 24. | Carpenter, W.; Bens, E. M. Tetrahedron 1970, 26, 59. doi:10.1016/0040-4020(70)85007-4 |
| 25. | Winberg, H. E.; Downing, J. R.; Coffman, D. D. J. Am. Chem. Soc. 1965, 87, 2054. doi:10.1021/ja01087a039 |
| 26. | Wiberg, N.; Buchler, J. W. Angew. Chem., Int. Ed. Engl. 1962, 1, 406. doi:10.1002/anie.196204061 |
| 27. | Wiberg, N.; Buchler, J. W. Chem. Ber. 1963, 96, 3223. doi:10.1002/cber.19630961218 |
| 28. | Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 1998, 63, 5385. doi:10.1021/jo980201+ |
| 29. | Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M.; Ndedi, A. Tetrahedron Lett. 1998, 39, 8853. doi:10.1016/S0040-4039(98)02028-0 |
| 30. | Burkholder, C.; Dolbier, W. R., Jr.; Médebielle, M.; Aït-Mohand, S. Tetrahedron Lett. 2001, 42, 3077. doi:10.1016/S0040-4039(01)00388-4 |
| 31. | Médebielle, M.; Keirouz, R.; Okada, E.; Ashida, T. Synlett 2001, 821. doi:10.1055/s-2001-14594 |
| 32. | Aït-Mohand, S.; Takechi, N.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2001, 3, 4271. doi:10.1021/ol016933x |
| 33. | Takechi, N.; Aït-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Tetrahedron Lett. 2002, 43, 4317. doi:10.1016/S0040-4039(02)00800-6 |
| 34. | Takechi, N.; Aït-Mohand, S.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 4671. doi:10.1021/ol0270374 |
| 35. | Médebielle, M.; Kato, K.; Dolbier, W. R., Jr. Tetrahedron Lett. 2003, 44, 7871. doi:10.1016/j.tetlet.2003.09.020 |
| 36. | Médebielle, M.; Hohn, S.; Okada, E.; Myoken, H.; Shibata, D. Tetrahedron Lett. 2005, 46, 7817. doi:10.1016/j.tetlet.2005.09.018 |
| 37. | Prakash, G. K. S.; Wang, Y.; Hu, J.; Olah, G. A. J. Fluorine Chem. 2005, 126, 1361. doi:10.1016/j.jfluchem.2005.07.011 |
| 38. | Xu, W.; Dolbier, W. R., Jr. J. Org. Chem. 2005, 70, 4741. doi:10.1021/jo050483v |
| 39. | Peng, W.; Zhao, J.; He, P.; Zhu, S. Synlett 2006, 296. doi:10.1055/s-2006-926225 |
| 40. | Peng, W.; Zhao, J.; Zhu, S. J. Fluorine Chem. 2006, 127, 360. doi:10.1016/j.jfluchem.2005.12.017 |
| 41. | Peng, W.; Zhao, J.; Zhu, S. Synthesis 2006, 1470. doi:10.1055/s-2006-926436 |
| 42. | Pooput, C.; Dolbier, W. R., Jr.; Médebielle, M. J. Org. Chem. 2006, 71, 3564. doi:10.1021/jo060250j |
| 43. | Médebielle, M.; Keirouz, R.; Okada, E.; Shibata, D.; Dolbier, W. R., Jr. Tetrahedron Lett. 2008, 49, 589. doi:10.1016/j.tetlet.2007.11.146 |
| 44. | Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2003, 44, 6433. doi:10.1016/S0040-4039(03)01594-6 |
| 45. | Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2004, 45, 5121. doi:10.1016/j.tetlet.2004.04.166 |
| 46. | Giuglio-Tonolo, G.; Terme, T.; Vanelle, P. Synlett 2005, 251. doi:10.1055/s-2004-837227 |
| 47. | Amiri-Attou, O.; Terme, T.; Vanelle, P. Synlett 2005, 3047. doi:10.1055/s-2005-921916 |
| 48. | Nishiyama, Y.; Kawabata, H.; Kobayashi, A.; Nishino, T.; Sonoda, N. Tetrahedron Lett. 2005, 46, 867. doi:10.1016/j.tetlet.2004.11.114 |
| 49. | Montana, M.; Crozet, M. D.; Castera-Ducros, C.; Terme, T.; Vanelle, P. Heterocycles 2008, 75, 925. |
| 50. | Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2006, 47, 6573. doi:10.1016/j.tetlet.2006.07.030 |
| 51. | Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2005, 46, 8373. doi:10.1016/j.tetlet.2005.09.152 |
| 52. | Nishiyama, Y.; Kobayashi, A. Tetrahedron Lett. 2006, 47, 5565. doi:10.1016/j.tetlet.2006.05.141 |
| 53. | Amiri-Attou, O.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2008, 49, 1016. doi:10.1016/j.tetlet.2007.12.011 |
| 54. | Kuroboshi, M.; Goto, K.; Mochizuki, M.; Tanaka, H. Synlett 1999, 1930. doi:10.1055/s-1999-2995 |
| 55. | Kuroboshi, M.; Tanaka, M.; Kishimoto, S.; Goto, K.; Mochizuki, M.; Tanaka, H. Tetrahedron Lett. 2000, 41, 81. doi:10.1016/S0040-4039(99)02006-7 |
| 56. | Kuroboshi, M.; Waki, Y.; Tanaka, H. Synlett 2002, 637. doi:10.1055/s-2002-22719 |
| 57. | Kuroboshi, M.; Waki, Y.; Tanaka, H. J. Org. Chem. 2003, 68, 3938. doi:10.1021/jo0207473 |
| 58. | Park, S. B.; Alper, H. Tetrahedron Lett. 2004, 45, 5515. doi:10.1016/j.tetlet.2004.05.013 |
| 59. | Kuroboshi, M.; Takeda, T.; Motoki, R.; Tanaka, H. Chem. Lett. 2005, 34, 530. doi:10.1246/cl.2005.530 |
| 71. | Bommezijn, S.; Martin, C. G.; Kennedy, A. R.; Lizos, D.; Murphy, J. A. Org. Lett. 2001, 3, 3405. doi:10.1021/ol0166449 |
| 10. | Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995. doi:10.1039/a900335e |
| 66. | Murphy, J. A.; Scott, K. A.; Sinclair, R. S.; Martin, C. G.; Kennedy, A. R.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 2000, 2395. doi:10.1039/b002565h |
| 67. | Murphy, J. A.; Scott, K. A.; Sinclair, R. S.; Lewis, N. Tetrahedron Lett. 1997, 38, 7295. doi:10.1016/S0040-4039(97)01695-X |
| 5. | Bashir, N.; Patro, B.; Murphy, J. A. In Advances in Free Radical Chemistry, Vol. 2; Zard, S. Z., Ed.; JAI Press: Stamford, CT, U.S.A., 1999; pp 123–150. |
| 6. | Murphy, J. A. The Radical-Polar Crossover Reaction. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 298–315. doi:10.1002/9783527618293.ch15 |
| 7. | Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Chem. Commun. 1993, 295. doi:10.1039/C39930000295 |
| 8. | Fletcher, R. J.; Lampard, C.; Murphy, J. A.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1995, 623. doi:10.1039/P19950000623 |
| 9. | Murphy, J. A.; Rasheed, F.; Gastaldi, S.; Ravishanker, T.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 1997, 1549. doi:10.1039/a607060d |
| 10. | Callaghan, O.; Lampard, C.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 995. doi:10.1039/a900335e |
| 11. | Lampard, C.; Murphy, J. A.; Rasheed, F.; Lewis, N.; Hursthouse, M. B.; Hibbs, D. E. Tetrahedron Lett. 1994, 35, 8675. doi:10.1016/S0040-4039(00)78469-3 |
| 12. | Murphy, J. A.; Rasheed, F.; Roome, S. J.; Lewis, N. Chem. Commun. 1996, 737. doi:10.1039/cc9960000737 |
| 13. | Fletcher, R.; Kizil, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. J. Chem. Soc., Perkin Trans. 1 1998, 2341. doi:10.1039/a802974a |
| 14. | Kizil, M.; Lampard, C.; Murphy, J. A. Tetrahedron Lett. 1996, 37, 2511. doi:10.1016/0040-4039(96)00306-1 |
| 15. | Fletcher, R. J.; Hibbs, D. E.; Hursthouse, M.; Lampard, C.; Murphy, J. A.; Roome, S. J. Chem. Commun. 1996, 739. doi:10.1039/cc9960000739 |
| 16. | Koizumi, T.; Bashir, N.; Murphy, J. A. Tetrahedron Lett. 1997, 38, 7635. doi:10.1016/S0040-4039(97)01813-3 |
| 17. | Koizumi, T.; Bashir, N.; Kennedy, A. R.; Murphy, J. A. J. Chem. Soc., Perkin Trans. 1 1999, 3637. doi:10.1039/a906684e |
| 18. | LeStrat, F.; Murphy, J. A.; Hughes, M. Org. Lett. 2002, 4, 2735. doi:10.1021/ol0262643 |
| 66. | Murphy, J. A.; Scott, K. A.; Sinclair, R. S.; Martin, C. G.; Kennedy, A. R.; Lewis, N. J. Chem. Soc., Perkin Trans. 1 2000, 2395. doi:10.1039/b002565h |
| 68. | Freidlina, R. Kh.; Terent’ev, A. B. Adv. Free-Radical Chem. 1980, 6, 1. |
| 69. | Freidlina, R. Kh. Adv. Free-Radical Chem. 1965, 1, 211. |
| 53. | Amiri-Attou, O.; Terme, T.; Médebielle, M.; Vanelle, P. Tetrahedron Lett. 2008, 49, 1016. doi:10.1016/j.tetlet.2007.12.011 |
| 51. | Montana, M.; Terme, T.; Vanelle, P. Tetrahedron Lett. 2005, 46, 8373. doi:10.1016/j.tetlet.2005.09.152 |
| 52. | Nishiyama, Y.; Kobayashi, A. Tetrahedron Lett. 2006, 47, 5565. doi:10.1016/j.tetlet.2006.05.141 |
| 62. | Ashton, P. R.; Balzani, V.; Becher, J.; Credi, A.; Fyfe, M. C. T.; Mattersteig, G.; Menzer, S.; Nielsen, M. B.; Raymo, F. M.; Stoddart, J. F.; Venturi, M.; Willliams, D. J. J. Am. Chem. Soc. 1999, 121, 3951. doi:10.1021/ja984341c |
| 63. |
Simonet, J.; Pilard, J.-F. Electrogenerated Reagents. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker Inc.: New York, 1991; pp 1163–1225.
See in particular pp 1171 ff. |
| 61. | Pause, L.; Robert, M.; Savéant, J.-M. J. Am. Chem. Soc. 1999, 121, 7158. doi:10.1021/ja991365q |
| 60. | Andrieux, C. P.; Pinson, J. J. Am. Chem. Soc. 2003, 125, 14801. doi:10.1021/ja0374574 |
| 60. | Andrieux, C. P.; Pinson, J. J. Am. Chem. Soc. 2003, 125, 14801. doi:10.1021/ja0374574 |
| 60. | Andrieux, C. P.; Pinson, J. J. Am. Chem. Soc. 2003, 125, 14801. doi:10.1021/ja0374574 |
| 54. | Kuroboshi, M.; Goto, K.; Mochizuki, M.; Tanaka, H. Synlett 1999, 1930. doi:10.1055/s-1999-2995 |
| 55. | Kuroboshi, M.; Tanaka, M.; Kishimoto, S.; Goto, K.; Mochizuki, M.; Tanaka, H. Tetrahedron Lett. 2000, 41, 81. doi:10.1016/S0040-4039(99)02006-7 |
| 56. | Kuroboshi, M.; Waki, Y.; Tanaka, H. Synlett 2002, 637. doi:10.1055/s-2002-22719 |
| 57. | Kuroboshi, M.; Waki, Y.; Tanaka, H. J. Org. Chem. 2003, 68, 3938. doi:10.1021/jo0207473 |
| 58. | Park, S. B.; Alper, H. Tetrahedron Lett. 2004, 45, 5515. doi:10.1016/j.tetlet.2004.05.013 |
| 59. | Kuroboshi, M.; Takeda, T.; Motoki, R.; Tanaka, H. Chem. Lett. 2005, 34, 530. doi:10.1246/cl.2005.530 |
© 2009 Mahesh et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)