Abstract
Simple, straightforward and optimized procedures for preparing extended π-conjugated linkers are described. Either unsubstituted or 4-donor substituted π-linkers bearing a styryl, biphenyl, phenylethenylphenyl, and phenylethynylphenyl π-conjugated backbone are functionalized with boronic pinacol esters as well as with terminal acetylene moieties allowing their further use as building blocks in Suzuki–Miyaura or Sonogashira coupling reactions.

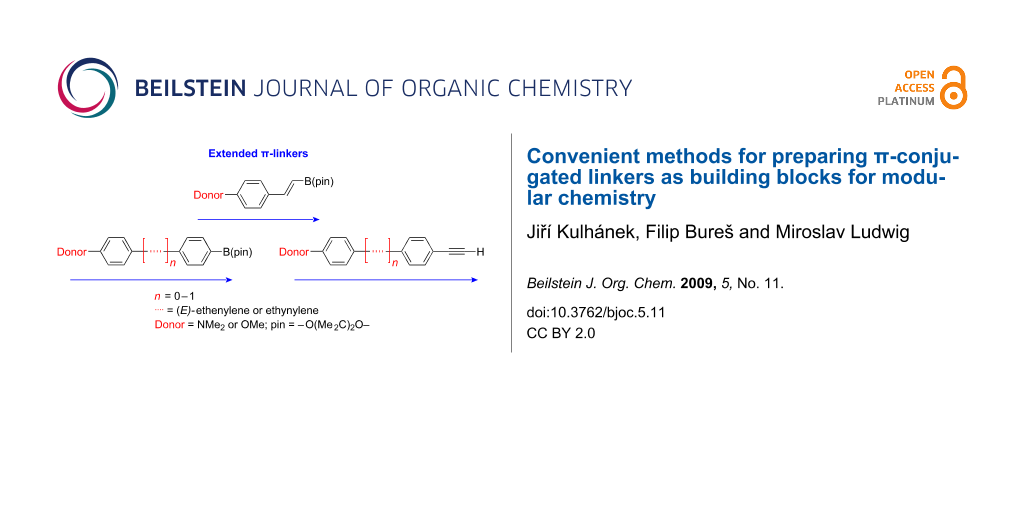
Graphical Abstract
Introduction
Development of new organic compounds with improved and advanced properties is one of the most important goals of modern material chemistry. Organic chemists steadily attempt to design and synthesize novel and well-defined organic push-pull systems with prospective applications as chromophores for nonlinear optics (NLO) [1-5], dyes [6], electronic and photonic devices [7,8], organic light-emitting diodes (OLED) [9] or functional polymers [10-13]. A typical push-pull chromophore consists of a polar A-π-D system with a planar π-system end-capped by a strong electron donor (D) and a strong electron acceptor (A). The π-conjugated system ensuring charge-transfer (CT) between the donor (D = NR2, OR groups etc.) and the acceptor (A = NO2, CN groups etc.) is most commonly comprised of double and triple bonds, aromatic and heteroaromatic rings as well as their combinations [14-19]. A typical synthetic approach to CT chromophores involves either a stepwise formation of the target molecule [19,20] or a separate preparation of the donor as well as the acceptor moieties and their final combination [21,22]. It is already well known that the HOMO/LUMO gap and polarizability of the push-pull systems with the given donors and acceptors can be finely tailored by the extension or shortening of the π-conjugated path between the donor and acceptor [19,21,23-25]. Thus, the latter modular synthetic approach seems to be more suitable for the property tuning described above. The final combination, C–C bond formation, of the donor and acceptor chromophore moieties is usually accomplished by cross-coupling reactions, in particular by the Suzuki–Miyaura [26,27] or the Sonogashira [28] reactions. Consequently, the availability of the suitably substituted π-conjugated linkers of various lengths bearing boronic ester functionality or terminal acetylene is crucial for such a synthetic approach. Hence, we report here a convenient synthesis as well as characterization of either unsubstituted (R = H) or donor substituted (R = NMe2, OMe) π-conjugated linkers designed for the Suzuki–Miyaura and Sonogashira cross-couplings with a systematically varied and enlarged π-conjugated path (Figure 1).
![[1860-5397-5-11-1]](/bjoc/content/figures/1860-5397-5-11-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Basic and newly proposed π-conjugated linkers designed for the Suzuki–Miyaura and Sonogashira cross-coupling.
Figure 1: Basic and newly proposed π-conjugated linkers designed for the Suzuki–Miyaura and Sonogashira cross...
Whereas the simplest linkers such as 4-substituted phenylboronic pinacol esters 1a–c [29] and ethynylbenzenes 2a–c [30] are well known and also commercially available, the proposed dioxaborolanes 3–6 feature styryl (series 3), biphenylyl (series 4), (E)-phenylethenylphenyl (series 5), and phenylethynylphenyl (series 6) linkers and H (series a), OMe (series b), and NMe2 (series c) groups as the substituent R, respectively (Figure 1, Table 1). The terminal acetylenes 7c–9c possess only the strongest NMe2 donor and have an identical backbone to the one described above (Figure 1, Table 1).
Table 1: Optimized synthetic procedures and yields for the preparation of 3–9.
| Entry | Product (R) | Method | Yield (%) |
|---|---|---|---|
| 1 | 3c (NMe2) | A | 73a |
| 2 | 4b (OMe) | B | 81a |
| 3 | 4c (NMe2) | B | 83a |
| 4 | 5a (H) | C | 76b |
| 5 | 5b (OMe) | C | 69a |
| 6 | 5c (NMe2) | C | 82a |
| 7 | 6a (H) | D | 78a |
| 8 | 6b (OMe) | D | 72a |
| 9 | 6c (NMe2) | D | 91a |
| 10 | 10/7c | E | 98/92c |
| 11 | 11/8c | E | 97/91c |
| 12 | 12/9c | E | 99/89c |
aYield of the final coupling step. bHorner–Wadsworth–Emmons reaction. cYield of the Sonogashira cross-coupling (10–12) and final TMS-group removal to the terminal acetylenes 7c–9c.
Results and Discussion
Synthesis of boronic pinacol esters 3–6
Whilst two styryl dioxaborolanes 3a–b are known [31,32] and commercially available, the N,N-dimethylamino substituted derivative 3c needed to be synthesized. In order to achieve pure (E)-3c, at first, a hydroboration of the commercially available terminal acetylene 2c with catecholborane was examined. Despite all attempts to optimize the reaction conditions, 3c could not be prepared this way and was not even detected in the crude reaction mixture. Thus the above hydroboration reported by Perner and co-workers [33] proved to be infeasible. However, in light of the report by Itami and Yoshida [34], we attempted the Mizoroki–Heck C–H arylation of 4-bromo-N,N-dimethylaniline with an equimolar amount of vinylboronate pinacol ester leading to the desired 3c in 73% yield (Scheme 1, Method A).
![[1860-5397-5-11-i1]](/bjoc/content/inline/1860-5397-5-11-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Convenient synthetic methods leading to π-linkers 3–6.
Scheme 1: Convenient synthetic methods leading to π-linkers 3–6.
4-Substituted 4′-bromobiphenyl intermediates necessary for the preparation of 4a–c were synthesized by the Suzuki–Miyaura cross-coupling of 1-bromo-4-iodobenzene with the corresponding boronic acids/esters 1a–c in the yields of 82, 84, and 91%, respectively. A routine procedure involving a lithiation and reaction with triisopropyl borate followed by esterification with pinacol afforded dioxaborolanes 4b–c in the yields of 81 and 83% (Scheme 1, Method B). The biphenyl-4-boronic acid corresponding to 4a was also commercially available while 4b was reported as a side product [35] without full characterization.
Whereas 5a was easily accessible as a pure (E)-product from the pinacol ester of 4-formylphenylboronic acid and diethyl benzylphosphonate through the Horner–Wadsworth–Emmons reaction in 76% yield [36], methoxy and N,N-dimethylamino substituted (E)-4-bromostilbenes were synthesized from the corresponding benzaldehydes and 4-bromobenzyl(triphenyl)phosphonium bromide [37] by the Wittig reaction [37,38] in 37 and 54% yields, respectively. In contrast to the Horner–Wadsworth–Emmons reaction, this procedure afforded both (E)- and (Z)-stilbenes that were isomerized by heating with traces of iodine in toluene to afford pure (E)-stilbenes [38]. However, compared to N,N-dimethylaminostilbene, the isomerization of 4-bromo-4′-methoxystilbene required twelve times the prolonged reaction time (4 vs. 48 h) while the isomerisation of unsubstituted 4-bromostilbene did not take a place at all. Hence, 5a had to be prepared by the Horner–Wadsworth–Emmons reaction described above. These substituted 4-bromostilbenes could be most effectively converted to target pinacol esters 5a–c via borylation (Scheme 1, Method C) utilizing bis(pinacolato)diboron (pin2B2) [39] in a mixed solvent system DMSO/dioxane ensuring good solubility. It should be noted here that 5a–c were also accessible via the routine sequence showed for Method B. On the contrary, borylation of 4-bromobiphenyls with pin2B2 (Method C) yielded only traces of 4a–c.
Linear phenylethynylphenyl π-linkers 6a–c were gained by a Sonogashira cross-coupling between the pinacol ester of 4-iodophenylboronic acid [39] and terminal acetylenes 2a–c (Scheme 1, Method D) in 72–91% yield. Table 1 (entries 1–9) summarizes the used synthetic methods and yields for the particular dioxaborolanes 3–6.
Synthesis of terminal acetylenes 7–9
Synthesis of N,N-dimethylamino substituted terminal acetylenes 7c–9c was accomplished by Sonogashira cross-coupling as shown on the Scheme 2 (Method E). The reaction utilizes the 4-bromo derivatives used as precursors for the construction of dioxaborolanes 4–6 and the product of the Sonogashira cross-coupling between 2c and 1,4-diiodobenzene (54% yield). Since the Sonogashira reaction between bromo derivatives and trimethylsilylacetylene proved to be sluggish and low yielding (even with a large excess of acetylene and elevated temperature), the bromo derivatives were converted to the corresponding iodo derivatives by lithiation and quenched with iodine (see Supporting Information File 1). Thus the Sonogashira reaction on iodo derivatives smoothly furnished TMS-protected acetylenes 10–12 in high yields and reaction times of about 30 min while the subsequent TMS group removal using TBAF (tetrabutylammonium fluoride) afforded desired π-linkers 7c–9c (Scheme 2, Table 1, entries 10–12). A similar synthesis of the linear π-linker 9c has already been reported [40]. Moreover, unprotected terminal acetylenes 7c–9c showed good stability with no signs of decomposition upon standing over several months which facilitate their storage and use.
![[1860-5397-5-11-i2]](/bjoc/content/inline/1860-5397-5-11-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Sonogashira cross-coupling leading to π-linkers 7c–9c.
Scheme 2: Sonogashira cross-coupling leading to π-linkers 7c–9c.
Conclusion
It has been shown that extended, donor-substituted π-conjugated linkers can be easily prepared using commercially available precursors under either conventional or modern synthetic conditions. The reaction procedures reported here refer to the optimized procedures for each class of derivatives. Overall 12 extended π-linkers have been easily synthesized (8 of them are new compounds) utilizing procedures such as a lithiation/reaction with triisopropyl borate/esterification with pinacol, Mizoroki–Heck coupling with vinylboronate pinacol ester, borylation with bis(pinacolato)diboron or Sonogashira coupling. Further application of the above boronic esters as well as terminal acetylenes for the construction of imidazole-based D-π-A systems by Suzuki–Miyaura and Sonogashira reaction is currently in progress in our laboratory.
References
-
Albota, M.; Beljonne, D.; Brédas, J.-L.; Ehrlich, J. E.; Fu, J.-Y.; Heikal, A. A.; Hess, S. E.; Kogej, T.; Levin, M. D.; Marder, S. R.; McCord-Maughon, D.; Perry, J. W.; Röckel, H.; Rumi, M.; Subramaniam, G.; Webb, W. W.; Wu, X.-L.; Xu, C. Science 1998, 281, 1653–1656. doi:10.1126/science.281.5383.1653
Return to citation in text: [1] -
Marder, S. R. Chem. Commun. 2006, 131–134. doi:10.1039/b512646k
Return to citation in text: [1] -
Kivala, M.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Gross, M.; Diederich, F. Angew. Chem., Int. Ed. 2007, 46, 6357–6360. doi:10.1002/anie.200701733
Return to citation in text: [1] -
Bureš, F.; Schweizer, W. B.; Boudon, C.; Gisselbrecht, J.-P.; Gross, M.; Diederich, F. Eur. J. Org. Chem. 2008, 994–1004. doi:10.1002/ejoc.200700970
Return to citation in text: [1] -
Juríček, M.; Kasák, P.; Stach, M.; Putala, M. Tetrahedron Lett. 2007, 48, 8869–8873. doi:10.1016/j.tetlet.2007.10.044
Return to citation in text: [1] -
Li, Z.; Qin, A.; Lam, J. W. Y.; Dong, Y.; Dong, Y.; Ye, C.; Williams, I. D.; Tang, B. Z. Macromolecules 2006, 39, 1436–1442. doi:10.1021/ma051924f
Return to citation in text: [1] -
Cumpston, B. H.; Ananthavel, S. P.; Barlow, S.; Dyer, D. L.; Ehrlich, J. E.; Erskine, L. L.; Heikal, A. A.; Kuebler, S. M.; Lee, I.-Y. S.; McCord-Maughon, D.; Qin, J.; Röckel, H.; Rumi, M.; Wu, X.-L.; Marder, S. R.; Perry, J. W. Nature 1999, 398, 51–54. doi:10.1038/17989
Return to citation in text: [1] -
Moonen, N. N. P.; Gist, R.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Kawai, T.; Kishioka, A.; Gross, M.; Irie, M.; Diederich, F. Org. Biomol. Chem. 2003, 1, 2032–2034. doi:10.1039/b303879c
Return to citation in text: [1] -
Haldi, A.; Kimyonok, A.; Domercq, B.; Hayden, L. E.; Jones, S. C.; Marder, S. R.; Weck, M.; Kippelen, B. Adv. Funct. Mater. 2008, 18, 3056–3062. doi:10.1002/adfm.200800446
Return to citation in text: [1] -
Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem., Int. Ed. 1998, 37, 402–428. doi:10.1002/(SICI)1521-3773(19980302)37:4<402::AID-ANIE402>3.0.CO;2-9
Return to citation in text: [1] -
Ma, H.; Liu, S.; Luo, J.; Suresh, S.; Liu, L.; Kang, S. H.; Haller, M.; Sassa, T.; Dalton, L. R.; Jen, A. K.-Y. Adv. Funct. Mater. 2002, 12, 565–574. doi:10.1002/1616-3028(20020916)12:9<565::AID-ADFM565>3.0.CO;2-8
Return to citation in text: [1] -
Kivala, M.; Diederich, F. Acc. Chem. Res. 2009, 42, 235–248. doi:10.1021/ar8001238
Return to citation in text: [1] -
Do, J. Y.; Ju, J. J. Macromol. Chem. Phys. 2005, 206, 1326–1331. doi:10.1002/macp.200400547
Return to citation in text: [1] -
Pahadi, N. K.; Camacho, D. H.; Nakamura, I.; Yamamoto, Y. J. Org. Chem. 2006, 71, 1152–1155. doi:10.1021/jo052262v
Return to citation in text: [1] -
Hennrich, G.; Asselberghs, I.; Clays, K.; Persoons, A. J. Org. Chem. 2004, 69, 5077–5081. doi:10.1021/jo049279i
Return to citation in text: [1] -
Wang, J.; Lu, M.; Pan, Y.; Peng, Z. J. Org. Chem. 2002, 67, 7781–7786. doi:10.1021/jo026186x
Return to citation in text: [1] -
Kivala, M.; Stanoeva, T.; Michinobu, T.; Frank, B.; Gescheidt, G.; Diederich, F. Chem.–Eur. J. 2008, 14, 7638–7647. doi:10.1002/chem.200800716
Return to citation in text: [1] -
Spitler, E. L.; Shirtcliff, L. D.; Haley, M. H. J. Org. Chem. 2007, 72, 86–96. doi:10.1021/jo061712w
Return to citation in text: [1] -
Patel, A.; Bureš, F.; Ludwig, M.; Kulhánek, J.; Pytela, O.; Růžička, A. Heterocycles 2009, 78, 999–1013. doi:10.3987/COM-08-11609
Return to citation in text: [1] [2] [3] -
Batista, R. M. F.; Costa, S. P. G.; Belsley, M.; Lodeiro, C.; Raposo, M. M. M. Tetrahedron 2008, 64, 9230–9238. doi:10.1016/j.tet.2008.07.043
Return to citation in text: [1] -
Bureš, F.; Schweizer, W. B.; May, J. C.; Boudon, C.; Gisselbrecht, J.-P.; Gross, M.; Biaggio, I.; Diederich, F. Chem.–Eur. J. 2007, 13, 5378–5387. doi:10.1002/chem.200601735
Return to citation in text: [1] [2] -
Andersson, A. S.; Kerndrup, L.; Madsen, A. Ø.; Kilså, K.; Nielsen, M. B.; La Porta, P. R.; Biaggio, I. J. Org. Chem. 2009, 74, 375–382. doi:10.1021/jo802190q
Return to citation in text: [1] -
May, J. C.; Biaggio, I.; Bureš, F.; Diederich, F. Appl. Phys. Lett. 2007, 90, No. 251106. doi:10.1063/1.2750396
Return to citation in text: [1] -
Wan, J.-H.; Feng, J.-C.; Wen, G.-A.; Wei, W.; Fan, Q.-L.; Wang, C.-M.; Wang, H.-Y.; Zhu, R.; Yuan, X.-D.; Huang, C.-H.; Huang, W. J. Org. Chem. 2006, 71, 2565–2571. doi:10.1021/jo0521596
Return to citation in text: [1] -
Bureš, F.; Pytela, O.; Diederich, F. J. Phys. Org. Chem. 2009, 22, 155–162. doi:10.1002/poc.1443
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633–9695. doi:10.1016/S0040-4020(02)01188-2
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Murata, M.; Watanabe, S.; Masuda, Y. J. Org. Chem. 1997, 62, 6458–6459. doi:10.1021/jo970963p
Return to citation in text: [1] -
Elangovan, A.; Wang, Y.-H.; Ho, T.-I. Org. Lett. 2003, 5, 1841–1844. doi:10.1021/ol034320+
Return to citation in text: [1] -
Mkhalid, I. A. I.; Coapes, R. B.; Edes, S. N.; Coventry, D. N.; Souza, F. E. S.; Thomas, R. L.; Hall, J. J.; Bi, S.-W.; Lin, Z.; Marder, T. B. Dalton Trans. 2008, 1055–1064. doi:10.1039/b715584k
Return to citation in text: [1] -
Murata, M.; Kawakita, K.; Asana, T.; Watanabe, S.; Masuda, Y. Bull. Chem. Soc. Jpn. 2002, 75, 825–829. doi:10.1246/bcsj.75.825
Return to citation in text: [1] -
Perner, R. J.; Lee, C.-H.; Jiang, M.; Gu, Y.-G.; DiDomenico, S.; Bayburt, E. K.; Alexander, K. M.; Kohlhaas, K. L.; Jarvis, M. F.; Kowaluk, E. L.; Bhagwat, S. S. Bioorg. Med. Chem. Lett. 2005, 15, 2803–2807. doi:10.1016/j.bmcl.2005.03.098
Return to citation in text: [1] -
Itami, K.; Tonogaki, K.; Ohashi, Y.; Yoshida, J.-i. Org. Lett. 2004, 6, 4093–4096. doi:10.1021/ol048217b
Return to citation in text: [1] -
Chaumeil, H.; Le Drian, C.; Defoin, A. Synthesis 2002, 757–760. doi:10.1055/s-2002-25773
Return to citation in text: [1] -
Oehlke, A.; Auer, A. A.; Jahre, I.; Walfort, B.; Rüffer, T.; Zoufalá, P.; Lang, H.; Spange, S. J. Org. Chem. 2007, 72, 4328–4339. doi:10.1021/jo070084v
Return to citation in text: [1] -
Ren, Y.; Yu, X.-Q.; Zhang, D.-J.; Wang, D.; Zhang, M.-L.; Xu, G.-B.; Zhao, X.; Tian, Y.-P.; Shao, Z.-S.; Jiang, M.-H. J. Mater. Chem. 2002, 12, 3431–3437. doi:10.1039/b206578a
Return to citation in text: [1] [2] -
Liu, Z.-q.; Fang, Q.; Wang, D.; Cao, D.-x.; Xue, G.; Yu, W.-t.; Lei, H. Chem.–Eur. J. 2003, 9, 5074–5084. doi:10.1002/chem.200304833
Return to citation in text: [1] [2] -
Perttu, E. K.; Arnold, M.; Iovine, P. M. Tetrahedron Lett. 2005, 46, 8753–8756. doi:10.1016/j.tetlet.2005.10.033
Return to citation in text: [1] [2] -
Rodriguez, J. G.; Esquivias, J.; Lafuente, A.; Rubio, L. Tetrahedron 2006, 62, 3112–3122. doi:10.1016/j.tet.2006.01.032
Return to citation in text: [1]
| 37. | Ren, Y.; Yu, X.-Q.; Zhang, D.-J.; Wang, D.; Zhang, M.-L.; Xu, G.-B.; Zhao, X.; Tian, Y.-P.; Shao, Z.-S.; Jiang, M.-H. J. Mater. Chem. 2002, 12, 3431–3437. doi:10.1039/b206578a |
| 35. | Chaumeil, H.; Le Drian, C.; Defoin, A. Synthesis 2002, 757–760. doi:10.1055/s-2002-25773 |
| 36. | Oehlke, A.; Auer, A. A.; Jahre, I.; Walfort, B.; Rüffer, T.; Zoufalá, P.; Lang, H.; Spange, S. J. Org. Chem. 2007, 72, 4328–4339. doi:10.1021/jo070084v |
| 1. | Albota, M.; Beljonne, D.; Brédas, J.-L.; Ehrlich, J. E.; Fu, J.-Y.; Heikal, A. A.; Hess, S. E.; Kogej, T.; Levin, M. D.; Marder, S. R.; McCord-Maughon, D.; Perry, J. W.; Röckel, H.; Rumi, M.; Subramaniam, G.; Webb, W. W.; Wu, X.-L.; Xu, C. Science 1998, 281, 1653–1656. doi:10.1126/science.281.5383.1653 |
| 2. | Marder, S. R. Chem. Commun. 2006, 131–134. doi:10.1039/b512646k |
| 3. | Kivala, M.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Gross, M.; Diederich, F. Angew. Chem., Int. Ed. 2007, 46, 6357–6360. doi:10.1002/anie.200701733 |
| 4. | Bureš, F.; Schweizer, W. B.; Boudon, C.; Gisselbrecht, J.-P.; Gross, M.; Diederich, F. Eur. J. Org. Chem. 2008, 994–1004. doi:10.1002/ejoc.200700970 |
| 5. | Juríček, M.; Kasák, P.; Stach, M.; Putala, M. Tetrahedron Lett. 2007, 48, 8869–8873. doi:10.1016/j.tetlet.2007.10.044 |
| 10. | Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem., Int. Ed. 1998, 37, 402–428. doi:10.1002/(SICI)1521-3773(19980302)37:4<402::AID-ANIE402>3.0.CO;2-9 |
| 11. | Ma, H.; Liu, S.; Luo, J.; Suresh, S.; Liu, L.; Kang, S. H.; Haller, M.; Sassa, T.; Dalton, L. R.; Jen, A. K.-Y. Adv. Funct. Mater. 2002, 12, 565–574. doi:10.1002/1616-3028(20020916)12:9<565::AID-ADFM565>3.0.CO;2-8 |
| 12. | Kivala, M.; Diederich, F. Acc. Chem. Res. 2009, 42, 235–248. doi:10.1021/ar8001238 |
| 13. | Do, J. Y.; Ju, J. J. Macromol. Chem. Phys. 2005, 206, 1326–1331. doi:10.1002/macp.200400547 |
| 33. | Perner, R. J.; Lee, C.-H.; Jiang, M.; Gu, Y.-G.; DiDomenico, S.; Bayburt, E. K.; Alexander, K. M.; Kohlhaas, K. L.; Jarvis, M. F.; Kowaluk, E. L.; Bhagwat, S. S. Bioorg. Med. Chem. Lett. 2005, 15, 2803–2807. doi:10.1016/j.bmcl.2005.03.098 |
| 9. | Haldi, A.; Kimyonok, A.; Domercq, B.; Hayden, L. E.; Jones, S. C.; Marder, S. R.; Weck, M.; Kippelen, B. Adv. Funct. Mater. 2008, 18, 3056–3062. doi:10.1002/adfm.200800446 |
| 34. | Itami, K.; Tonogaki, K.; Ohashi, Y.; Yoshida, J.-i. Org. Lett. 2004, 6, 4093–4096. doi:10.1021/ol048217b |
| 7. | Cumpston, B. H.; Ananthavel, S. P.; Barlow, S.; Dyer, D. L.; Ehrlich, J. E.; Erskine, L. L.; Heikal, A. A.; Kuebler, S. M.; Lee, I.-Y. S.; McCord-Maughon, D.; Qin, J.; Röckel, H.; Rumi, M.; Wu, X.-L.; Marder, S. R.; Perry, J. W. Nature 1999, 398, 51–54. doi:10.1038/17989 |
| 8. | Moonen, N. N. P.; Gist, R.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Kawai, T.; Kishioka, A.; Gross, M.; Irie, M.; Diederich, F. Org. Biomol. Chem. 2003, 1, 2032–2034. doi:10.1039/b303879c |
| 30. | Elangovan, A.; Wang, Y.-H.; Ho, T.-I. Org. Lett. 2003, 5, 1841–1844. doi:10.1021/ol034320+ |
| 40. | Rodriguez, J. G.; Esquivias, J.; Lafuente, A.; Rubio, L. Tetrahedron 2006, 62, 3112–3122. doi:10.1016/j.tet.2006.01.032 |
| 6. | Li, Z.; Qin, A.; Lam, J. W. Y.; Dong, Y.; Dong, Y.; Ye, C.; Williams, I. D.; Tang, B. Z. Macromolecules 2006, 39, 1436–1442. doi:10.1021/ma051924f |
| 31. | Mkhalid, I. A. I.; Coapes, R. B.; Edes, S. N.; Coventry, D. N.; Souza, F. E. S.; Thomas, R. L.; Hall, J. J.; Bi, S.-W.; Lin, Z.; Marder, T. B. Dalton Trans. 2008, 1055–1064. doi:10.1039/b715584k |
| 32. | Murata, M.; Kawakita, K.; Asana, T.; Watanabe, S.; Masuda, Y. Bull. Chem. Soc. Jpn. 2002, 75, 825–829. doi:10.1246/bcsj.75.825 |
| 19. | Patel, A.; Bureš, F.; Ludwig, M.; Kulhánek, J.; Pytela, O.; Růžička, A. Heterocycles 2009, 78, 999–1013. doi:10.3987/COM-08-11609 |
| 21. | Bureš, F.; Schweizer, W. B.; May, J. C.; Boudon, C.; Gisselbrecht, J.-P.; Gross, M.; Biaggio, I.; Diederich, F. Chem.–Eur. J. 2007, 13, 5378–5387. doi:10.1002/chem.200601735 |
| 23. | May, J. C.; Biaggio, I.; Bureš, F.; Diederich, F. Appl. Phys. Lett. 2007, 90, No. 251106. doi:10.1063/1.2750396 |
| 24. | Wan, J.-H.; Feng, J.-C.; Wen, G.-A.; Wei, W.; Fan, Q.-L.; Wang, C.-M.; Wang, H.-Y.; Zhu, R.; Yuan, X.-D.; Huang, C.-H.; Huang, W. J. Org. Chem. 2006, 71, 2565–2571. doi:10.1021/jo0521596 |
| 25. | Bureš, F.; Pytela, O.; Diederich, F. J. Phys. Org. Chem. 2009, 22, 155–162. doi:10.1002/poc.1443 |
| 28. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 39. | Perttu, E. K.; Arnold, M.; Iovine, P. M. Tetrahedron Lett. 2005, 46, 8753–8756. doi:10.1016/j.tetlet.2005.10.033 |
| 21. | Bureš, F.; Schweizer, W. B.; May, J. C.; Boudon, C.; Gisselbrecht, J.-P.; Gross, M.; Biaggio, I.; Diederich, F. Chem.–Eur. J. 2007, 13, 5378–5387. doi:10.1002/chem.200601735 |
| 22. | Andersson, A. S.; Kerndrup, L.; Madsen, A. Ø.; Kilså, K.; Nielsen, M. B.; La Porta, P. R.; Biaggio, I. J. Org. Chem. 2009, 74, 375–382. doi:10.1021/jo802190q |
| 29. | Murata, M.; Watanabe, S.; Masuda, Y. J. Org. Chem. 1997, 62, 6458–6459. doi:10.1021/jo970963p |
| 39. | Perttu, E. K.; Arnold, M.; Iovine, P. M. Tetrahedron Lett. 2005, 46, 8753–8756. doi:10.1016/j.tetlet.2005.10.033 |
| 19. | Patel, A.; Bureš, F.; Ludwig, M.; Kulhánek, J.; Pytela, O.; Růžička, A. Heterocycles 2009, 78, 999–1013. doi:10.3987/COM-08-11609 |
| 20. | Batista, R. M. F.; Costa, S. P. G.; Belsley, M.; Lodeiro, C.; Raposo, M. M. M. Tetrahedron 2008, 64, 9230–9238. doi:10.1016/j.tet.2008.07.043 |
| 37. | Ren, Y.; Yu, X.-Q.; Zhang, D.-J.; Wang, D.; Zhang, M.-L.; Xu, G.-B.; Zhao, X.; Tian, Y.-P.; Shao, Z.-S.; Jiang, M.-H. J. Mater. Chem. 2002, 12, 3431–3437. doi:10.1039/b206578a |
| 38. | Liu, Z.-q.; Fang, Q.; Wang, D.; Cao, D.-x.; Xue, G.; Yu, W.-t.; Lei, H. Chem.–Eur. J. 2003, 9, 5074–5084. doi:10.1002/chem.200304833 |
| 14. | Pahadi, N. K.; Camacho, D. H.; Nakamura, I.; Yamamoto, Y. J. Org. Chem. 2006, 71, 1152–1155. doi:10.1021/jo052262v |
| 15. | Hennrich, G.; Asselberghs, I.; Clays, K.; Persoons, A. J. Org. Chem. 2004, 69, 5077–5081. doi:10.1021/jo049279i |
| 16. | Wang, J.; Lu, M.; Pan, Y.; Peng, Z. J. Org. Chem. 2002, 67, 7781–7786. doi:10.1021/jo026186x |
| 17. | Kivala, M.; Stanoeva, T.; Michinobu, T.; Frank, B.; Gescheidt, G.; Diederich, F. Chem.–Eur. J. 2008, 14, 7638–7647. doi:10.1002/chem.200800716 |
| 18. | Spitler, E. L.; Shirtcliff, L. D.; Haley, M. H. J. Org. Chem. 2007, 72, 86–96. doi:10.1021/jo061712w |
| 19. | Patel, A.; Bureš, F.; Ludwig, M.; Kulhánek, J.; Pytela, O.; Růžička, A. Heterocycles 2009, 78, 999–1013. doi:10.3987/COM-08-11609 |
| 26. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 27. | Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633–9695. doi:10.1016/S0040-4020(02)01188-2 |
| 38. | Liu, Z.-q.; Fang, Q.; Wang, D.; Cao, D.-x.; Xue, G.; Yu, W.-t.; Lei, H. Chem.–Eur. J. 2003, 9, 5074–5084. doi:10.1002/chem.200304833 |
© 2009 Kulhánek et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)