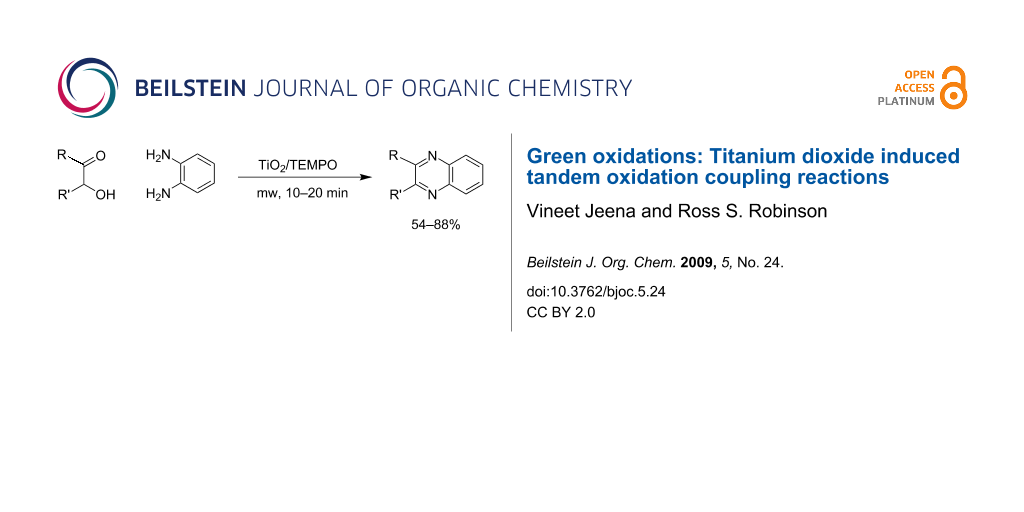
Abstract
The application of titanium dioxide as an oxidant in tandem oxidation type processes is described. Under microwave irradiation, quinoxalines have been synthesized in good yields from the corresponding α-hydroxyketones.

Graphical Abstract
Introduction
Titanium dioxide has found widespread industrial application ranging from whiteners in paint [1], and additives in food [2], to UV absorbers in sunscreen lotions [3]. Its popularity stems from its inertness, low cost, and chemical stability under irradiation [4]. In the laboratory, its utility has been extended to the photodegradation of pesticides [5] and carcinogenic dyes [6], to sterilization against bacteria [7]. From a synthetic chemistry point of view, titanium dioxide’s main use has been to oxidize alcohols to its corresponding carbonyl derivatives which has been reported many times [8-10]. Titanium dioxide has also been used to synthesize dihydropyrazines [11], piperazines [12], and quinoxalines [13] although in low yields.
Herein, we describe the application of titanium dioxide in conjunction with 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) as an oxidant in the synthesis of quinoxalines, via the tandem oxidation process (TOP) (Scheme 1).
![[1860-5397-5-24-i1]](/bjoc/content/inline/1860-5397-5-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Titanium dioxide/TEMPO mediated synthesis of quinoxalines.
Scheme 1: Titanium dioxide/TEMPO mediated synthesis of quinoxalines.
Results and Discussion
Titanium dioxide in the anatase phase is a photocatalyst with a band gap of 3.2 eV corresponding to a wavelength of 387 nm [14]. Thus, we attempted to evaluate titanium dioxide as a potential TOP type catalyst based on the excellent work of Taylor and co-workers [15,16] by examining its behaviour under various energy sources (Table 1).
Table 1: Evaluation of energy sources.
![[Graphic 1]](/bjoc/content/inline/1860-5397-5-24-i2.svg?max-width=637&scale=1.0)
|
|||
| Entry | Energy source | Time (h) | Yield (%)a,b |
|---|---|---|---|
| i | Natural sunlight | 3 | 90 |
| ii | Incandescent lamp | 3 | 25 |
| iii | UV light | 3 | 10 |
| iv | Microwave | 10 min | 87c |
aMixture of TiO2 (0.5 mmol), alcohol (0.5 mmol), amine (0.5 mmol) and TEMPO (0.05 mmol) in 3 ml methanol. bIsolated yield. cAbsence of solvent.
Titanium dioxide is known to be highly reactive under sunlight [17] and this seemed to be the most obvious starting point. Initially, 2-hydroxyacetophenone was reacted with o-phenylenediamine under natural sunlight. We were delighted to observe the formation of the quinoxaline derivative in an isolated yield of 90% in 3 h. While encouraged by this result we immediately realized the limitations of this procedure. The use of an incandescent lamp gave a disappointing yield of 25% while a low wattage UV light afforded an equally disappointing yield of 10% in 3 h. We explored the use of microwave energy, which has been claimed to substantially improve reaction rates [18]. Surprisingly, the use of microwave energy produced the best result with the quinoxaline derivative isolated in a yield of 87% in 10 min. With the optimized procedure in hand, an investigation into the scope of the methodology, using a range of alcohols and diamines, was conducted (Table 2).
Table 2: Titanium dioxide catalysed tandem oxidation process under microwave irradiation.
| Entry | α-Hydroxyketone | Diamine | Quinoxaline | Time (min) | Yield (%)a |
|---|---|---|---|---|---|
| i |
1a
![[Graphic 2]](/bjoc/content/inline/1860-5397-5-24-i3.svg?max-width=637&scale=1.0)
|
2a
![[Graphic 3]](/bjoc/content/inline/1860-5397-5-24-i4.svg?max-width=637&scale=1.0)
|
3a
![[Graphic 4]](/bjoc/content/inline/1860-5397-5-24-i5.svg?max-width=637&scale=1.0)
|
10 | 87 |
| ii |
1b
![[Graphic 5]](/bjoc/content/inline/1860-5397-5-24-i6.svg?max-width=637&scale=1.0)
|
2a
![[Graphic 6]](/bjoc/content/inline/1860-5397-5-24-i7.svg?max-width=637&scale=1.0)
|
3b
![[Graphic 7]](/bjoc/content/inline/1860-5397-5-24-i8.svg?max-width=637&scale=1.0)
|
10 | 60 |
| iii |
1c
![[Graphic 8]](/bjoc/content/inline/1860-5397-5-24-i9.svg?max-width=637&scale=1.0)
|
2a
![[Graphic 9]](/bjoc/content/inline/1860-5397-5-24-i10.svg?max-width=637&scale=1.0)
|
3c
![[Graphic 10]](/bjoc/content/inline/1860-5397-5-24-i11.svg?max-width=637&scale=1.0)
|
20 | 81 |
| iv |
1d
![[Graphic 11]](/bjoc/content/inline/1860-5397-5-24-i12.svg?max-width=637&scale=1.0)
|
2a
![[Graphic 12]](/bjoc/content/inline/1860-5397-5-24-i13.svg?max-width=637&scale=1.0)
|
3d
![[Graphic 13]](/bjoc/content/inline/1860-5397-5-24-i14.svg?max-width=637&scale=1.0)
|
10 | 88 |
| v |
1a
![[Graphic 14]](/bjoc/content/inline/1860-5397-5-24-i15.svg?max-width=637&scale=1.0)
|
2b
![[Graphic 15]](/bjoc/content/inline/1860-5397-5-24-i16.svg?max-width=637&scale=1.0)
|
3e
![[Graphic 16]](/bjoc/content/inline/1860-5397-5-24-i17.svg?max-width=637&scale=1.0)
|
10 | 83b |
| vi |
1c
![[Graphic 17]](/bjoc/content/inline/1860-5397-5-24-i18.svg?max-width=637&scale=1.0)
|
2b
![[Graphic 18]](/bjoc/content/inline/1860-5397-5-24-i19.svg?max-width=637&scale=1.0)
|
3f
![[Graphic 19]](/bjoc/content/inline/1860-5397-5-24-i20.svg?max-width=637&scale=1.0)
|
20 | 54 |
| vii |
1d
![[Graphic 20]](/bjoc/content/inline/1860-5397-5-24-i21.svg?max-width=637&scale=1.0)
|
2b
![[Graphic 21]](/bjoc/content/inline/1860-5397-5-24-i22.svg?max-width=637&scale=1.0)
|
3g
![[Graphic 22]](/bjoc/content/inline/1860-5397-5-24-i23.svg?max-width=637&scale=1.0)
|
20 | 56 |
aIsolated yield. bIsolated as a mixture of regioisomers.
After the successful synthesis of quinoxaline 3a, the effect of an alkyl substituent was explored, which is often claimed to be problematic due to the ‘hyper-reactivity’ of the intermediate keto-aldehyde [19]. The reaction afforded the quinoxaline derivative 3b in a satisfactory isolated yield of 60%. The coupling of secondary alcohols 1c and 1d both proceeded smoothly, even though the hindered alcohol 1c required a longer reaction time. Finally, the diamine component was varied (entries v-vii) with satisfactory yields obtained for both primary and secondary alcohols.
Concerning the mechanism, when titanium dioxide is irradiated with an appropriate energy source, electrons are promoted from the valence band to the conduction band leaving behind positive holes in the valence band [20]. The positive holes and electrons migrate to the surface where the holes react with water (or bound hydroxyl groups) to produce hydroxyl radicals which are strong oxidants [21]. Nevertheless, the activation of titanium dioxide under microwave irradiation is surprising since UV light is more intense than microwave energy. However, low levels of hydroxyl radical formation have been reported when titanium dioxide was subjected to microwave irradiation [22]. It has been suggested [23] that microwave energy can couple with a crystalline solid generating a non-thermal distribution, resulting in an increase in ion mobility, which leads to the diffusion of electrons and positive holes to the surface and subsequent hydroxyl radical formation [24]. It is believed that a similar interaction is taking place in our system and in conjunction with TEMPO lead to the formation of keto-aldehydes or diketones, which are trapped in situ to produce the required products.
In an attempt to extend the methodology, a preliminary investigation into a titanium dioxide mediated tandem Wittig reaction was attempted in which the intermediate aldehyde is trapped by a stabilised phosphorane. Unfortunately, NMR spectroscopic analysis revealed a multitude of peaks, none of which were attributable to the product. It is believed that this is due to the non-selective nature of the hydroxyl radicals [25]. Currently studies are under way to moderate the reactivity of the titanium dioxide. In a recent publication [26], Zhao and co-workers reported the use of dye-sensitized titanium dioxide and TEMPO for the selective oxidation of alcohols to aldehydes and ketones. In our studies we have been examining similar systems with a view to incorporating them into tandem type processes.
Conclusion
In summary, we have reported the use of titanium dioxide as a tandem oxidation catalyst, demonstrated by the rapid synthesis of quinoxalines under microwave irradiation. The process is advantageous due to the green credentials and low cost of the oxidant, short reaction times and good yields.
Experimental
2-Phenylquinoxaline (3a)
2-Hydroxyacetophenone (0.068 g, 0.50 mmol), o-phenylenediamine (0.054 g, 0.50 mmol), titanium dioxide (0.040 g, 0.50 mmol) and TEMPO (0.008 g, 0.050 mmol) were added to a sealed 10 mL CEM Discover® reaction vial equipped with a magnetic stirrer bar. The reaction vial was irradiated (at 150 W with cooling) for 10 min (2 × 5 min) at 150 °C, after which the vessel was rapidly cooled to 50 °C by the unit. The reaction mixture was diluted with dichloromethane (DCM) and passed through a short silica plug. The solvent was removed in vacuo to produce a crude product which was purified using radial chromatography (3:1 PE:EtOAc) to afford the pure product.
Supporting Information
Supporting information features experimental procedures and spectroscopic analysis for compounds 3a–3g coupling reactions.
| Supporting Information File 1: Experimental and spectroscopic data for: Green oxidations: Titanium dioxide induced tandem oxidation | ||
| Format: DOC | Size: 740.0 KB | Download |
References
-
Tryk, D. A.; Fujishima, A.; Honda, K. Electrochim. Acta 2000, 45, 2363–2376. doi:10.1016/S0013-4686(00)00337-6
Return to citation in text: [1] -
Phillips, L. Y.; Barbano, D. M. J. Dairy Sci. 1997, 80, 2726–2733.
Return to citation in text: [1] -
Hewitt, J. P. Cosmet. Toiletries 1999, 114, 59–63.
Return to citation in text: [1] -
Palmisano, G.; Augugliaro, V.; Pagliaro, M.; Palmisano, L. Chem. Commun. 2007, 3425–3437. doi:10.1039/b700395c
Return to citation in text: [1] -
Mahalakshmi, M.; Arabindoo, B.; Palanichamy, M.; Murugesan, V. J. Hazard. Mater. 2007, 143, 240–245. doi:10.1016/j.jhazmat.2006.09.008
Return to citation in text: [1] -
Abu Tariq, M.; Faisal, M.; Muneer, M. J. Hazard. Mater. 2005, 127, 172–179. doi:10.1016/j.jhazmat.2005.07.001
Return to citation in text: [1] -
Yu, J. C.; Xie, Y.; Tang, H. Y.; Zhang, L.; Chan, H. C.; Zhao, J. J. Photochem. Photobiol., A: Chem. 2003, 156, 235–241. doi:10.1016/S1010-6030(03)00008-X
Return to citation in text: [1] -
Mohamed, O. S.; Gaber, A. E.-A. M.; Abdel-Wahab, A. A. J. Photochem. Photobiol., A: Chem. 2002, 148, 205–210. doi:10.1016/S1010-6030(02)00044-8
Return to citation in text: [1] -
Hussein, F. H.; Rudham, R. J. Chem. Soc., Faraday Trans. 1 1987, 83, 1631–1639. doi:10.1039/F19878301631
Return to citation in text: [1] -
Gassim, F. A.-Z. G.; Alkhateeb, A. N.; Hussein, F. H. Desalination 2007, 209, 342–349. doi:10.1016/j.desal.2007.04.049
Return to citation in text: [1] -
Subba Rao, K. V.; Srinivas, B.; Prasad, A. R.; Subrahmanyam, M. Chem. Commun. 2000, 1533–1534. doi:10.1039/b003934i
Return to citation in text: [1] -
Subba Rao, K. V.; Subrahmanyam, M. Photochem. Photobiol. Sci. 2002, 1, 597–599. doi:10.1039/b203431j
Return to citation in text: [1] -
Subba Rao, K. V.; Subrahmanyam, M. Chem. Lett. 2002, 31, 234–235. doi:10.1246/cl.2002.234
Return to citation in text: [1] -
Wunderlich, W.; Oekermann, T.; Miao, L.; Hue, N. T.; Tanemura, S.; Tanemura, M. J. Ceram. Process. Res. 2004, 5, 343–354.
Return to citation in text: [1] -
Raw, S. A.; Wilfred, C. D.; Taylor, R. J. K. Org. Biomol. Chem. 2004, 2, 788–796. doi:10.1039/b315689c
Return to citation in text: [1] -
Robinson, R. S.; Taylor, R. J. K. Synlett 2005, 1003–1005. doi:10.1055/s-2005-864830
Return to citation in text: [1] -
Fujishima, A.; Rao, T. N.; Tryk, D. A. J. Photochem. Photobiol., C: Photochem. Rev. 2000, 1, 1–21. doi:10.1016/S1389-5567(00)00002-2
Return to citation in text: [1] -
Herrero, M. A.; Kremsner, J. M.; Kappe, C. O. J. Org. Chem. 2008, 73, 36–47. doi:10.1021/jo7022697
Return to citation in text: [1] -
Ireland, R. E.; Norbeck, D. W. J. Org. Chem. 1985, 50, 2198–2200. doi:10.1021/jo00212a041
Return to citation in text: [1] -
Alberici, R. M.; Jardim, W. F. Appl. Catal., B: Environ. 1997, 14, 55–68. doi:10.1016/S0926-3373(97)00012-X
Return to citation in text: [1] -
Emeline, A. V.; Frolov, A. V.; Ryabchuk, V. K.; Serpone, N. J. Phys. Chem. B 2003, 107, 7109–7119. doi:10.1021/jp030126t
Return to citation in text: [1] -
Horikoshi, S.; Hidaka, H.; Serpone, N. Chem. Phys. Lett. 2003, 376, 475–480. doi:10.1016/S0009-2614(03)01007-8
Return to citation in text: [1] -
Booske, J. H.; Cooper, R. F.; Dobson, I. J. Mater. Res. 1992, 7, 495–501. doi:10.1557/JMR.1992.0495
Return to citation in text: [1] -
Horikoshi, S.; Hidaka, H.; Serpone, N. J. Photochem. Photobiol., A: Chem. 2003, 159, 289–300. doi:10.1016/S1010-6030(03)00185-0
Return to citation in text: [1] -
Yurdakal, S.; Palmisano, G.; Loddo, V.; Augugliaro, V.; Palmisaono, L. J. Am. Chem. Soc. 2008, 130, 1568–1569. doi:10.1021/ja709989e
Return to citation in text: [1] -
Zhang, M.; Chen, C.; Ma, M.; Zhao, J. Angew. Chem., Int. Ed. 2008, 47, 9730–9733. doi:10.1002/anie.200803630
Return to citation in text: [1]
| 22. | Horikoshi, S.; Hidaka, H.; Serpone, N. Chem. Phys. Lett. 2003, 376, 475–480. doi:10.1016/S0009-2614(03)01007-8 |
| 20. | Alberici, R. M.; Jardim, W. F. Appl. Catal., B: Environ. 1997, 14, 55–68. doi:10.1016/S0926-3373(97)00012-X |
| 21. | Emeline, A. V.; Frolov, A. V.; Ryabchuk, V. K.; Serpone, N. J. Phys. Chem. B 2003, 107, 7109–7119. doi:10.1021/jp030126t |
| 1. | Tryk, D. A.; Fujishima, A.; Honda, K. Electrochim. Acta 2000, 45, 2363–2376. doi:10.1016/S0013-4686(00)00337-6 |
| 5. | Mahalakshmi, M.; Arabindoo, B.; Palanichamy, M.; Murugesan, V. J. Hazard. Mater. 2007, 143, 240–245. doi:10.1016/j.jhazmat.2006.09.008 |
| 18. | Herrero, M. A.; Kremsner, J. M.; Kappe, C. O. J. Org. Chem. 2008, 73, 36–47. doi:10.1021/jo7022697 |
| 4. | Palmisano, G.; Augugliaro, V.; Pagliaro, M.; Palmisano, L. Chem. Commun. 2007, 3425–3437. doi:10.1039/b700395c |
| 19. | Ireland, R. E.; Norbeck, D. W. J. Org. Chem. 1985, 50, 2198–2200. doi:10.1021/jo00212a041 |
| 15. | Raw, S. A.; Wilfred, C. D.; Taylor, R. J. K. Org. Biomol. Chem. 2004, 2, 788–796. doi:10.1039/b315689c |
| 16. | Robinson, R. S.; Taylor, R. J. K. Synlett 2005, 1003–1005. doi:10.1055/s-2005-864830 |
| 17. | Fujishima, A.; Rao, T. N.; Tryk, D. A. J. Photochem. Photobiol., C: Photochem. Rev. 2000, 1, 1–21. doi:10.1016/S1389-5567(00)00002-2 |
| 11. | Subba Rao, K. V.; Srinivas, B.; Prasad, A. R.; Subrahmanyam, M. Chem. Commun. 2000, 1533–1534. doi:10.1039/b003934i |
| 13. | Subba Rao, K. V.; Subrahmanyam, M. Chem. Lett. 2002, 31, 234–235. doi:10.1246/cl.2002.234 |
| 25. | Yurdakal, S.; Palmisano, G.; Loddo, V.; Augugliaro, V.; Palmisaono, L. J. Am. Chem. Soc. 2008, 130, 1568–1569. doi:10.1021/ja709989e |
| 8. | Mohamed, O. S.; Gaber, A. E.-A. M.; Abdel-Wahab, A. A. J. Photochem. Photobiol., A: Chem. 2002, 148, 205–210. doi:10.1016/S1010-6030(02)00044-8 |
| 9. | Hussein, F. H.; Rudham, R. J. Chem. Soc., Faraday Trans. 1 1987, 83, 1631–1639. doi:10.1039/F19878301631 |
| 10. | Gassim, F. A.-Z. G.; Alkhateeb, A. N.; Hussein, F. H. Desalination 2007, 209, 342–349. doi:10.1016/j.desal.2007.04.049 |
| 14. | Wunderlich, W.; Oekermann, T.; Miao, L.; Hue, N. T.; Tanemura, S.; Tanemura, M. J. Ceram. Process. Res. 2004, 5, 343–354. |
| 26. | Zhang, M.; Chen, C.; Ma, M.; Zhao, J. Angew. Chem., Int. Ed. 2008, 47, 9730–9733. doi:10.1002/anie.200803630 |
| 7. | Yu, J. C.; Xie, Y.; Tang, H. Y.; Zhang, L.; Chan, H. C.; Zhao, J. J. Photochem. Photobiol., A: Chem. 2003, 156, 235–241. doi:10.1016/S1010-6030(03)00008-X |
| 23. | Booske, J. H.; Cooper, R. F.; Dobson, I. J. Mater. Res. 1992, 7, 495–501. doi:10.1557/JMR.1992.0495 |
| 6. | Abu Tariq, M.; Faisal, M.; Muneer, M. J. Hazard. Mater. 2005, 127, 172–179. doi:10.1016/j.jhazmat.2005.07.001 |
| 12. | Subba Rao, K. V.; Subrahmanyam, M. Photochem. Photobiol. Sci. 2002, 1, 597–599. doi:10.1039/b203431j |
| 24. | Horikoshi, S.; Hidaka, H.; Serpone, N. J. Photochem. Photobiol., A: Chem. 2003, 159, 289–300. doi:10.1016/S1010-6030(03)00185-0 |
© 2009 Jeena and Robinson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)