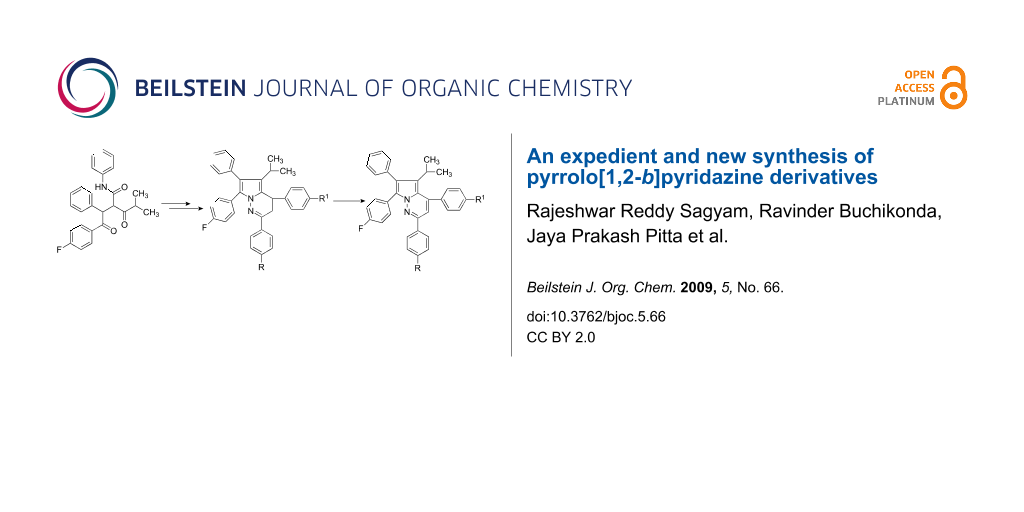
Abstract
The reaction of 2-[2-(4-fluorophenyl)-2-oxo-1-phenylethyl]-4-methyl-3-oxo-pentanoic acid phenylamide with tertiary butyl carbazate and subsequent condensation of the resulting carbamate derivative with a chalcone provided a facile new approach to pyrrolo[1,2-b]pyridazine derivatives.

Graphical Abstract
Introduction
Pyrrolopyridazine derivatives have various biological applications [1-8], and their fluorescent properties have been investigated for potential use in sensors, lasers, and semiconductor devices [9-13].
The synthesis and properties of pyrrolo[1,2-b]pyridazine derivatives were reviewed in 1977 by Kuhla and Lombardino [14]. Subsequently, new methods for the synthesis of these compounds have been described, which can be classified into two main approaches. The first involves condensation reactions, such as the condensation of oxazolo[3,2-b]pyridazinium perchlorates with malononitrile, ethyl cyanoacetate and ethyl malonate in the presence of sodium ethoxide [15]; the condensation of 1,4,7-triketones with hydrazine followed by dehydrogenation [16]; the condensation of cyanoacetic acid hydrazide with 3-bromo-1,1,3-tricyano-2-phenylpropene [17]; and the reaction between 3-chloropyridazines with propargylic alcohol in the presence of Pd(PPh3)2Cl2–CuI with diethylamine as the reaction medium [18,19]. The second approach is based on cycloaddition reactions, such as the cycloaddition of dimethyl acetylenedicarboxylate to the Reissert compound of pyridazine [20], the 1,3-dipolar cycloaddition of pyridazinium dichloromethylide generated by the carbene method [21], and the cycloaddition of alkylidene cyclopropane derivatives to pyridazine in the presence of Pd(PPh3)4 [22].
Pyrrolo[1,2-b]pyridazine derivatives can also be synthesized from 1-aminopyrrole and its derivatives. This original method was reported by Flitsch and Krämer (in 1968–9) [23,24], who obtained a series of unsubstituted pyrrolopyridazines from 1-aminopyrrole and β-dicarbonyl compounds. Benzoylacetone, on condensation with 1-aminopyrrole, forms only one isomer, 2-methyl-4-phenyl-pyrrolopyridazine, whereas benzoylacetaldehyde yields a mixture of 2-phenyl- and 4-phenyl-pyrrolopyridazine. 3-Phenylpyrrolopyridazine is obtained from phenylmalonaldehyde and 1-aminopyrrole [25]. Unsubstituted pyrrolopyridazine (Figure 1) was synthesized in 21% yield from 1-aminopyrrole and 3-ethoxyacrolein diethylacetal [26].
![[1860-5397-5-66-1]](/bjoc/content/figures/1860-5397-5-66-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Unsubstituted pyrrolo[1,2-b]pyridazine.
Figure 1: Unsubstituted pyrrolo[1,2-b]pyridazine.
As a part of our continued interest in the development of new synthetic methods for highly substituted pyrrole and indole derivatives [27,28], we have developed a new synthetic route to pyrrolo[1,2-b]pyridazines through a hitherto unprecedented approach from a BOC-protected 1-aminopyrrole derivative and α,β-unsaturated ketones.
Results and Discussion
2-[2-(4-Fluorophenyl)-2-oxo-1-phenylethyl]-4-methyl-3-oxo-pentanoic acid phenylamide 1 was reacted with tertiary butyl carbazate 2 in toluene and cyclohexane in the presence of p-toluenesulfonic acid (p-TSA) at reflux and the resulting [2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-phenylcarbamoyl-pyrrol-1-yl]-carbamic acid tert-butyl ester 3 was further condensed with 3-(4-fluorophenyl)-1-phenyl-propenone 4a [29] in the presence of p-TSA in the same medium. Pyrrolo[1,2-b]pyridazine derivatives, i.e. 4,7-bis-(4-fluorophenyl)-4a-isopropyl-2,6-diphenyl-4a,7-dihydropyrrolo[1,2-b]pyridazine-5-carboxylic acid phenylamide 5a, were expected as products in this synthetic sequence (Scheme 1). However, IR, mass, HRMS, and 1H, 13C, and 2D NMR spectral data of the product confirmed the structure of the product as 4,7-bis-(4-fluoro-phenyl)-5-isopropyl-2,6-diphenyl-3,4-dihydropyrrolo[1,2-b]pyridazine 6a (Table 1).
![[1860-5397-5-66-i1]](/bjoc/content/inline/1860-5397-5-66-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: i) p-TSA (0.5 equiv), toluene/cyclohexane (4:15), reflux, 15–18 h; ii) p-TSA (1.5 equiv), toluene/cyclohexane (30:20), reflux, 15–25 h.
Scheme 1: Reagents and conditions: i) p-TSA (0.5 equiv), toluene/cyclohexane (4:15), reflux, 15–18 h; ii) p-T...
In the mass spectrum of the compound, the molecular ion peak was observed at m/z 502 (M+), instead of m/z 621 (M+); HRMS data also confirmed the m/z 502 (M+) and molecular formula as C34H28F2N2, in accord with structure 6a and not with 5a. The IR spectrum lacked any –C=O absorption. The 1H NMR spectrum of 6a exhibited signals due to two methyl groups and the –CH of an isopropyl group at δ 0.85 (d, 3H), δ 1.15 (d, 3H), and δ 2.84–2.91 (m, 1H), respectively; –CH2 and –CH of pyridazine ring at δ 3.1–3.26 (ddd, 2H) and δ 4.71–4.74 (d, 1H), respectively; and aromatic protons at δ 6.88–7.63 (m, 18H). In the 13C NMR, the DEPT spectrum was characterized by the presence of signals due to 2 × CH3 and 1 × CH of isopropyl group; CH2 and CH of pyridazine ring at δ 22.6, 23.8, and 25.3; 31.5 and 34.7 ppm, respectively. Product 5a should exhibit a –C=O signal and no CH2 peak. In the DQCOSY spectrum, 1H–1H coupling between 2 × CH3 groups and –CH of the isopropyl group and also between the –CH2 and –CH groups of pyridazine ring was seen, but no coupling between the isopropyl group and the –CH2 and –CH of the pyridazine ring was observed. This indicates that the newly formed –CH2 and –CH are connected to each other, which is not possible in 5a. The HSQC spectrum exhibited 13C–1H coupling of two methyl groups at δ 23.0, 1.2 (d, 3H); δ 24.0, 0.90 (d, 3H) and –CH of isopropyl at δ 25.5, 2.85 (m, 1H), and also the –CH2 and –CH groups of pyridazine ring at δ 31.5, 3.1–3.3 (ddd, 2H); δ 35.0, 4.75 (d, 1H). The newly formed –CH2 is linked to a -CH group. It is reported [30] that the 4° carbons of the pyrrole ring resonate at 118 (C-8), 122 (C-6), 124 (C-5), and 132 (C-7). The HMBC spectrum displayed the 13C–1H correlations of 2 × CH3 with only C-5; iPr-CH with 2 × CH3, C-5, C-6, and C-8; CH of pyridazine ring (C-4) with C-3, C-8, C-5 (less), C-2, C-9, and C-10; CH2 (C-3) with C-4, C-2, C-8, C-5 (very small), C-13 (small), and C-9. These data strongly support the linkage of isopropyl group to C-5; C-4 to C-8, C-3; and C-3 to C-2, C-4. All these spectral data are in favor of 4,7-bis-(4-fluoro-phenyl)-5-isopropyl-2,6-diphenyl-3,4-dihydropyrrolo[1,2-b]pyridazine structure 6a (Figure 2), but not of 4,7-bis-(4-fluorophenyl)-4a-isopropyl-2,6-diphenyl-4a,7-dihydropyrrolo[1,2-b]pyridazine-5-carboxylic acid phenylamide 5a.
![[1860-5397-5-66-2]](/bjoc/content/figures/1860-5397-5-66-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Depiction with proprietary numbering of compound 6a.
Figure 2: Depiction with proprietary numbering of compound 6a.
A plausible mechanistic pathway for the formation of compounds 6a–j involves hydrolysis and decarboxylation of carbamate 3, subsequent condensation with chalcone 4a–j to provide alkenyl imine 9, its sequential hydrolysis and decarboxylation, followed by cyclization and migration of the isopropyl group (Scheme 2).
![[1860-5397-5-66-i2]](/bjoc/content/inline/1860-5397-5-66-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
To substantiate the proposed mechanism, the amine 8 was independently prepared from compound 3 by treatment with 33% hydrobromic acid in acetic acid at 30 °C followed by reaction with 4a in the presence of I2 (0.05 equiv) in refluxing ethyl alcohol to provide alkenyl imine 9, which was characterized on the basis of its mass, 1H and 13C NMR, DEPT, and IR spectral data. Compound 9 was heated in toluene in the presence of p-TSA for 10–12 h and the resulting compound was found to be identical to product 6a. Hydrolysis of the amide group and subsequent decarboxylation was carried out on pyrrole derivative 15 to afford the 2,3-diaryl pyrrole derivative 16 (Scheme 3).
![[1860-5397-5-66-i3]](/bjoc/content/inline/1860-5397-5-66-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: i) p-TSA (1.5 equiv), toluene/cyclohexane (1:1), reflux, 10.0 h.
Scheme 3: Reagents and conditions: i) p-TSA (1.5 equiv), toluene/cyclohexane (1:1), reflux, 10.0 h.
With a view to extending this protocol to aliphatic systems such as α,β-unsaturated ketones, carbamate 3 was treated with crotonaldehyde under similar conditions. However, the alkenyl imine analogue 9 thus obtained did not undergo further reaction. This may be due to the +I effect of alkyl groups, whereas in the case of aryl groups (−M effect) the olefinic carbon is electron deficient and therefore cyclization is favorable.
To aromatize the pyrrolopyridazine ring system, the compound 6a was heated in the presence of p-TSA in toluene at 110 °C for 25.0 h and the resulting compound to yield 4,7-bis-(4-fluorophenyl)-5-isopropyl-2,6-diphenylpyrrolo[1,2-b]pyridazine (17a, Scheme 4). Other pyrrolopyridazine derivatives 6a–j were converted into corresponding dehydro derivatives 17a–j under similar conditions (Table 2).
![[1860-5397-5-66-i4]](/bjoc/content/inline/1860-5397-5-66-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: i) p-TSA (2.0 equiv), toluene (30.0 volumes).
Scheme 4: Reagents and conditions: i) p-TSA (2.0 equiv), toluene (30.0 volumes).
Conclusion
In conclusion, a facile new approach has been developed for the synthesis of pyrrolo[1,2-b]pyridazine derivatives from commercially available and environmentally friendly chemicals. This newly developed method offers quick access to building blocks for various products with pyrrolo[1,2-b]pyridazine cores.
Experimental
The 1H, 13C NMR spectra were recorded in DMSO-d6 and CDCl3 at 200 or 400 MHz on a Mercury Plus/Varian Gemini 2000 FT NMR spectrometer. Proton chemical shifts (δ) were expressed in ppm with tetramethylsilane (TMS, δ 0.00) as internal standard. Spin multiplicities are given as s (singlet), d (doublet), t (triplet), and m (multiplet). FT-IR spectra were recorded in KBr dispersion with a Perkin-Elmer 1650 FT-IR spectrometer. Mass spectra (70 eV) were recorded on HP-5989 A LC-MS spectrometer. The high resolution mass spectroscopy (HRMS) analysis was performed on the Micromass LCT Premier XE mass spectrometer equipped with an ESI Lack spray source for accurate mass values (Water Corporation, Milford, MA, USA). Melting points were determined by the capillary method with a POLMON (Model MP-96) melting point apparatus. Solvent removal was accomplished by a Buchi rotary evaporator at house vacuum (30–40 Torr). Solvents and reagents were used without further purification. The purity of compounds was checked on silica gel coated aluminium plates (Merck).
(a) Procedure for compound 3: A mixture of 2-[2-(4-fluorophenyl)-2-oxo-1-phenylethyl]-4-methyl-3-oxo-pentanoic acid phenylamide (1, 5.0 g, 0.012 mol), tertiary butyl carbazate (2, 2.06 g, 0.0156 mol), and p-TSA (0.006 mol) in toluene (20.0 mL) and cyclohexane (75.0 mL) was maintained at reflux until no more water collected (reaction monitored by TLC). The reaction mixture was cooled to 30 °C, allowed to stand for 3 h, filtered and washed with cyclohexane (10.0 mL). The compound was washed again with cyclohexane (30.0 mL) and dried to give a white solid. mp 191–193 °C; 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.3 (d, 15H, 3CH3 of ester and 2CH3 of iPr), 3.1 (m, 1H, CH), 6.97–7.54 (m, 14H, Ar-H), 9.9 (s, 1H, NH amide), 10.3 (s, 1H, NH ester), both –NH groups were D2O exchangeable; IR KBr (cm−1): 3422, 3258, 1712, 1671; Anal. Calcd for C31H32FN3O3: C, 72.49; H, 6.28; N, 8.18. Found: C, 72.33; H, 6.42; N, 8.35.
(b) A typical procedure for compound 6a: A mixture of [2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-phenylcarbamoyl-pyrrol-1-yl]-carbamic acid tert-butyl ester (3, 5.0 g, 0.0097 mol), 3-(4-fluorophenyl)-1-phenyl-propenone (4a, 2.62 g, 0.0102 mol), and p-TSA (2.5 g, 0.0146 mol) in toluene (150.0 mL) and cyclohexane (100.0 mL) was maintained at reflux (azeotropic) for 19.0 h (reaction monitored by TLC). The reaction mixture was cooled to ambient temperature; ethyl acetate (40.0 mL) was added and washed first with water (25.0 mL) and then with 10% sodium bicarbonate solution (25.0 mL). The resulting organic layer was concentrated under vacuum and the crude product recrystallized from ethyl acetate (25.0 mL) to remove unreacted 9 (~5.0%). The resulting filtrate was concentrated under vacuum and further recrystallized from isopropyl alcohol (20.0 mL) to give a cream solid (Table 1). mp 203–205 °C; MS: m/z 502 (M+); HRMS data: m/z 502 (M+) and mol. formula: C34H28F2N2; 1H NMR (400 MHz, CDCl3, δ ppm): 0.85 (d, J = 7.2 Hz, 3H, CH3), 1.15 (d, J = 7.2 Hz, 3H, CH3), 2.84–2.91 (m, 1H, CH), 3.1–3.26 (ddd, J = 6.8, 16.4 Hz, 2H, CH2 of ring), 4.71–4.74 (d, J = 6.8 Hz, 1H, CH of ring), 6.88–7.63 (m, 18H, Ar-H); 13C NMR (100 MHz, CDCl3, δ ppm): 22.6, 23.8, 25.3, 31.5, 34.7, 114.0, 114.5, 115.2, 115.7, 118.4, 122.2, 124.0, 126.0, 126.2, 127.0, 127.7, 127.9, 128.3, 128.5, 129.7, 131.2, 132.50, 132.65, 136.1, 136.7, 139.5, 152.3, 159.0, 159.2, 163.8, 164.0; DEPT (200 MHz, CDCl3, δ ppm): CH3 carbons at δ 22.6, 23.8; aliphatic-CH carbons at δ 25.3, 34.7 and aromatic-CH carbons at δ 114.0–132.6; CH2 carbon at δ 31.5 ppm; DQCOSY (400 MHz, CDCl3, δ ppm, 1H–1H coupling): δ 0.8 and 1.2 coupled with δ 2.8 and δ 3.2 coupled with δ 4.8 ppm; HSQC (400 MHz, CDCl3, δ ppm, 13C–1H coupling): δ 23.0, 1.2 (d, 3H, CH3 of iPr); δ 24.0, 0.90 (d, 3H, CH3 of iPr); and δ 25.5, 2.85 (m, 1H, CH of iPr); δ 31.5, 3.1–3.3 (ddd, 2H, CH2 of pyridazine ring); δ 35.0, 4.75 (d, 1H, CH of pyridazine ring); and δ 113–134, 6.9–7.95 (m, 18H, Ar-H); the HMBC spectrum: 13C–1H correlations of 2 × CH3 (124, C-5); iPr-CH (2 × CH3, C-5, C-6, and C-8); CH of pyridazine ring (CH2, C-8, C-5 (less), C-2, C-9, and C-10); CH2 (C-4, C-8, C-5 (very less), C-13 (less), C-9, and C-2); IR KBr (cm−1): 3064, 1600, 1506, 1155; Anal. Calcd for C34H28F2N2: C, 81.25; H, 5.62; N, 5.57. Found: C, 81.41; H, 5.51; N, 5.74.
References
-
Ruxer, J. M.; Lachoux, C.; Ousset, J. B.; Torregrosa, J. L.; Mattioda, G. J. Heterocycl. Chem. 1994, 31, 409–417. doi:10.1002/jhet.5570310228
Return to citation in text: [1] -
Ungureanu, M.; Mangalagiu, I.; Grosu, G.; Petrovanu, M. Ann. Pharm. Fr. 1997, 55, 69–72.
Return to citation in text: [1] -
Nasir, A. I.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Kristensen, T.; Langhelle, B.; Bast, A.; Custers, I.; Haenen, G. R. M. M.; Wikström, H. Bioorg. Med. Chem. Lett. 1998, 8, 1829–1832. doi:10.1016/S0960-894X(98)00313-8
Return to citation in text: [1] -
Østby, O. B.; Dalhus, B.; Gundersen, L.-L.; Rise, F.; Bast, A.; Haenen, G. R. M. M. Eur. J. Org. Chem. 2000, 3763–3770. doi:10.1002/1099-0690(200011)2000:22<3763::AID-EJOC3763>3.0.CO;2-S
Return to citation in text: [1] -
Østby, O. B.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Fosnes, K.; Larsen, V.; Bast, A.; Custers, I.; Haenen, G. R. M. M. Arch. Pharm. 2001, 334, 21–24. doi:10.1002/1521-4184(200101)334:1<21::AID-ARDP21>3.0.CO;2-W
Return to citation in text: [1] -
Ohtani, M.; Fuji, M.; Fukui, Y.; Adachi, M. Pyrrolo[1,2-b]pyridazine Derivatives Having sPLA2 Inhibitory Effect. WO 059999, 1999.
Return to citation in text: [1] -
Salvati, M. E.; Illig, C. R.; Wilson, K. J.; Chen, J.; Meegalla, S. K.; Wall, M. J. Pyrrolopyridazine Compounds and Methods of Use Thereof for the Treatment of Proliferative Disorders. U.S. Patent 7,030,112, 2006.
Return to citation in text: [1] -
Fu, J.-M. Pyrrolo[1,2-b]pyridazine Compounds and Their Uses. U.S. Patent 7,074,791, 2006.
Return to citation in text: [1] -
Cheng, Y.; Ma, B.; Wudl, F. J. Mater. Chem. 1999, 9, 2183–2188. doi:10.1039/a903025e
Return to citation in text: [1] -
Mitsumori, T.; Bendikov, M.; Sedó, J.; Wudl, F. Chem. Mater. 2003, 15, 3759–3768. doi:10.1021/cm0340532
Return to citation in text: [1] -
Mitsumori, T.; Craig, I. M.; Martini, I. B.; Schwartz, B. J.; Wudl, F. Macromolecules 2005, 38, 4698–4704. doi:10.1021/ma048091y
Return to citation in text: [1] -
Swamy, K. M. K.; Park, M. S.; Han, S. J.; Kim, S. K.; Kim, J. H.; Lee, C.; Bang, H.; Kim, Y.; Kim, S.-J.; Yoon, J. Tetrahedron 2005, 61, 10227–10234. doi:10.1016/j.tet.2005.08.038
Return to citation in text: [1] -
Zbancioc, G. N.; Mangalagiu, I. I. Synlett 2006, 804–806. doi:10.1055/s-2006-932459
Return to citation in text: [1] -
Kuhla, D. E.; Lombardino, J. O. Adv. Heterocycl. Chem. 1977, 21, 1–63. doi:10.1016/S0065-2725(08)60729-1
Return to citation in text: [1] -
Satoh, K.; Miyasaka, T.; Arakawa, K. Yakugaku Zasshi 1977, 97, 422–429.
Return to citation in text: [1] -
Stetter, H.; Landscheidt, A. J. Heterocycl. Chem. 1979, 16, 839–843. doi:10.1002/jhet.5570160503
Return to citation in text: [1] -
Abdelrazek, F. M. Synth. Commun. 2005, 35, 2251–2258. doi:10.1080/00397910500184727
Return to citation in text: [1] -
Ohsawa, A.; Abe, Y.; Igeta, H. Bull. Chem. Soc. Jpn. 1980, 53, 3273–3275. doi:10.1246/bcsj.53.3273
Return to citation in text: [1] -
Ohsawa, A.; Abe, Y.; Igeta, H. Chem. Pharm. Bull. 1980, 28, 3488–3493.
Return to citation in text: [1] -
Khlebnikov, A. F.; Kostik, E. I.; Kostikov, R. R. Synthesis 1993, 568–570. doi:10.1055/s-1993-25905
Return to citation in text: [1] -
Veeraraghavan, S.; Bhattacharjee, D.; Popp, F. D. J. Heterocycl. Chem. 1981, 18, 443. doi:10.1002/jhet.5570180250
Return to citation in text: [1] -
Siriwardana, A. I.; Nakamura, I.; Yamamoto, Y. J. Org. Chem. 2004, 69, 3202–3204. doi:10.1021/jo035810i
Return to citation in text: [1] -
Flitsch, W.; Krämer, U. Tetrahedron Lett. 1968, 9, 1479–1484. doi:10.1016/S0040-4039(01)98983-X
Return to citation in text: [1] -
Flitsch, W.; Krämer, U.; Zimmermann, H. Chem. Ber. 1969, 102, 3268–3276. doi:10.1002/cber.19691021005
Return to citation in text: [1] -
Coppola, G. M.; Hardtmann, G. E.; Huegi, B. S. J. Heterocycl. Chem. 1974, 11, 51–56. doi:10.1002/jhet.5570110111
Return to citation in text: [1] -
Flitsch, W.; Krämer, U. Justus Liebigs Ann. Chem. 1970, 735, 35–46. doi:10.1002/jlac.19707350107
Return to citation in text: [1] -
Rajeshwar, R. S.; Pratap, R. P.; Mahesh, R. G.; Himabindu, V. J. Heterocycl. Chem. 2007, 44, 923–926. doi:10.1002/jhet.5570440429
Return to citation in text: [1] -
Rajeshwar, R. S.; Himabindu, V.; Pratap, R. P.; Katkam, S.; Mahesh, R. G. Synth. Commun., in press.
Return to citation in text: [1] -
Kohler, E. P.; Chadwell, H. M. Org. Synth. 1932, CV 1, 78–80.
Return to citation in text: [1] -
Abdolali, A.; Atieh, R.; Hamid, R. B. Synthesis 2008, 725–728. doi:10.1055/s-2008-1032168
Return to citation in text: [1]
| 1. | Ruxer, J. M.; Lachoux, C.; Ousset, J. B.; Torregrosa, J. L.; Mattioda, G. J. Heterocycl. Chem. 1994, 31, 409–417. doi:10.1002/jhet.5570310228 |
| 2. | Ungureanu, M.; Mangalagiu, I.; Grosu, G.; Petrovanu, M. Ann. Pharm. Fr. 1997, 55, 69–72. |
| 3. | Nasir, A. I.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Kristensen, T.; Langhelle, B.; Bast, A.; Custers, I.; Haenen, G. R. M. M.; Wikström, H. Bioorg. Med. Chem. Lett. 1998, 8, 1829–1832. doi:10.1016/S0960-894X(98)00313-8 |
| 4. | Østby, O. B.; Dalhus, B.; Gundersen, L.-L.; Rise, F.; Bast, A.; Haenen, G. R. M. M. Eur. J. Org. Chem. 2000, 3763–3770. doi:10.1002/1099-0690(200011)2000:22<3763::AID-EJOC3763>3.0.CO;2-S |
| 5. | Østby, O. B.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Fosnes, K.; Larsen, V.; Bast, A.; Custers, I.; Haenen, G. R. M. M. Arch. Pharm. 2001, 334, 21–24. doi:10.1002/1521-4184(200101)334:1<21::AID-ARDP21>3.0.CO;2-W |
| 6. | Ohtani, M.; Fuji, M.; Fukui, Y.; Adachi, M. Pyrrolo[1,2-b]pyridazine Derivatives Having sPLA2 Inhibitory Effect. WO 059999, 1999. |
| 7. | Salvati, M. E.; Illig, C. R.; Wilson, K. J.; Chen, J.; Meegalla, S. K.; Wall, M. J. Pyrrolopyridazine Compounds and Methods of Use Thereof for the Treatment of Proliferative Disorders. U.S. Patent 7,030,112, 2006. |
| 8. | Fu, J.-M. Pyrrolo[1,2-b]pyridazine Compounds and Their Uses. U.S. Patent 7,074,791, 2006. |
| 16. | Stetter, H.; Landscheidt, A. J. Heterocycl. Chem. 1979, 16, 839–843. doi:10.1002/jhet.5570160503 |
| 30. | Abdolali, A.; Atieh, R.; Hamid, R. B. Synthesis 2008, 725–728. doi:10.1055/s-2008-1032168 |
| 14. | Kuhla, D. E.; Lombardino, J. O. Adv. Heterocycl. Chem. 1977, 21, 1–63. doi:10.1016/S0065-2725(08)60729-1 |
| 26. | Flitsch, W.; Krämer, U. Justus Liebigs Ann. Chem. 1970, 735, 35–46. doi:10.1002/jlac.19707350107 |
| 9. | Cheng, Y.; Ma, B.; Wudl, F. J. Mater. Chem. 1999, 9, 2183–2188. doi:10.1039/a903025e |
| 10. | Mitsumori, T.; Bendikov, M.; Sedó, J.; Wudl, F. Chem. Mater. 2003, 15, 3759–3768. doi:10.1021/cm0340532 |
| 11. | Mitsumori, T.; Craig, I. M.; Martini, I. B.; Schwartz, B. J.; Wudl, F. Macromolecules 2005, 38, 4698–4704. doi:10.1021/ma048091y |
| 12. | Swamy, K. M. K.; Park, M. S.; Han, S. J.; Kim, S. K.; Kim, J. H.; Lee, C.; Bang, H.; Kim, Y.; Kim, S.-J.; Yoon, J. Tetrahedron 2005, 61, 10227–10234. doi:10.1016/j.tet.2005.08.038 |
| 13. | Zbancioc, G. N.; Mangalagiu, I. I. Synlett 2006, 804–806. doi:10.1055/s-2006-932459 |
| 27. | Rajeshwar, R. S.; Pratap, R. P.; Mahesh, R. G.; Himabindu, V. J. Heterocycl. Chem. 2007, 44, 923–926. doi:10.1002/jhet.5570440429 |
| 28. | Rajeshwar, R. S.; Himabindu, V.; Pratap, R. P.; Katkam, S.; Mahesh, R. G. Synth. Commun., in press. |
| 21. | Veeraraghavan, S.; Bhattacharjee, D.; Popp, F. D. J. Heterocycl. Chem. 1981, 18, 443. doi:10.1002/jhet.5570180250 |
| 23. | Flitsch, W.; Krämer, U. Tetrahedron Lett. 1968, 9, 1479–1484. doi:10.1016/S0040-4039(01)98983-X |
| 24. | Flitsch, W.; Krämer, U.; Zimmermann, H. Chem. Ber. 1969, 102, 3268–3276. doi:10.1002/cber.19691021005 |
| 20. | Khlebnikov, A. F.; Kostik, E. I.; Kostikov, R. R. Synthesis 1993, 568–570. doi:10.1055/s-1993-25905 |
| 25. | Coppola, G. M.; Hardtmann, G. E.; Huegi, B. S. J. Heterocycl. Chem. 1974, 11, 51–56. doi:10.1002/jhet.5570110111 |
| 18. | Ohsawa, A.; Abe, Y.; Igeta, H. Bull. Chem. Soc. Jpn. 1980, 53, 3273–3275. doi:10.1246/bcsj.53.3273 |
| 19. | Ohsawa, A.; Abe, Y.; Igeta, H. Chem. Pharm. Bull. 1980, 28, 3488–3493. |
| 17. | Abdelrazek, F. M. Synth. Commun. 2005, 35, 2251–2258. doi:10.1080/00397910500184727 |
| 22. | Siriwardana, A. I.; Nakamura, I.; Yamamoto, Y. J. Org. Chem. 2004, 69, 3202–3204. doi:10.1021/jo035810i |
© 2009 Sagyam et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)