Abstract
A series of dithienophenazines with different lengths of the oligomeric thiophene units (quaterthiophenes and sexithiophenes) was synthesized. The thiophene and phenazine units act as electron donors and acceptors, respectively, resulting in characteristic absorption spectra. The optical spectra were calculated using time-dependent density functional theory at the B3LYP/TZVP level and verify the experimental data. Adsorption of the dithienophenazines on highly ordered pyrolytic graphite (HOPG) was investigated by scanning tunneling microscopy, showing that one of the compounds forms highly organized self-assembled monolayers.
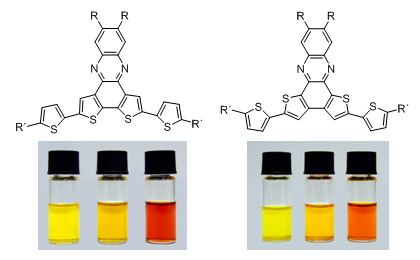
Graphical Abstract
Introduction
Thiophene based oligomers and polymers have drawn considerable interest as active materials in various fields of organic electronics such as organic light-emitting diodes (OLEDs), organic thin-film transistors (OTFTs), or organic photovoltaics (OPVs) [1-3]. Concerning applications, organic materials are highly attractive due to their low cost and their particularly simple deposition from either vacuum or solution (casting or printing), and thus they are already utilized in industrial manufacturing. Particularly, deposition from the solution phase requires sufficiently high solubility, and one approach to cover this aspect is the functionalization with long, flexible alkyl substituents. Additionally, in many cases (short) oligomers are considerably better soluble than their corresponding (long) polymers, and can, as a matter of principle, be obtained in higher purity with respect to defects/polymerization faults. Moreover, they act as model systems allowing estimation of the pure polymer properties by extrapolation of the oligomer properties to infinite molecular weight [4].
Beside pure thiophene-based (well-established) materials, compounds with additional electron-poor moieties are in focus, as they are known to shift the HOMO and LUMO levels towards lower energies, thus increasing the compounds' stability against oxidation. In addition, the HOMO-LUMO gap is reduced (by the donor-acceptor (DA) approach), thereby red-shifting the absorption edge. This is of special importance, as there is still a need for new materials (exhibiting additionally the above mentioned processability criteria) absorbing the longer wavelength region (> 600 nm) of the sunlight spectrum, being required for photocurrent generation also from respective low-energy photons and thus enhancing the overall light harvesting yield in organic photovoltaics (OPV). For example, in the field of fused N- and S-heterocycles, thieno[3,4-b]pyrazines with various side groups have already found applications in OPVs [5-8]. In addition, fused bithiophenes with an enforced planarity, and therefore reduced optical gaps, and an increased π–π overlap of the polycyclic molecule backbone have entered various fields of organic electronics [9-13].
Apart from their optoelectronic properties, linear and branched as well as cyclic and polycyclic thiophenes are valuable templates for the epitaxial coadsorption of adlayers, in particular for fullerenes and metallacycles [14-17].
Results and Discussion
Here we report the syntheses of two isomeric benzodithiophenediones, their respective phenazines and their coupling products with thiophene boronic acids. In both cases the thiophene units of the molecules act as electron rich parts (donors, D), whereas the phenazine moieties serve as electron deficient parts (acceptors, A), leading to a bathochromic shift of the UV–vis spectra in comparison with the non-condensed thiophene analogues. These assignments of the thiophene moieties as donors and the phenazine moieties as acceptors are confirmed by quantum-chemical calculations at the density-functional level.
Additionally, one of the compounds forms a self-assembled monolayer on a HOPG surface, as imaged by scanning tunneling microscopy (STM).
Synthesis
The synthesis of 4 is shown in Scheme 1. It is obtained in three steps from commercially available starting materials. The synthesis of 3 has previously been reported by benzoin condensation of the corresponding bithiophene derivative [18].
![[1860-5397-6-135-i1]](/bjoc/content/inline/1860-5397-6-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 4. a) 1) EtLi, Et2O, −78 °C, 1 h; 2) (COOMe)2, Et2O, −78 °C, 2 h, 33%; b) FeCl3, MeNO2, CH2Cl2, rt, 16 h, 71%; c) Br2, AcOH, CHCl3, reflux, 5 h, 95%.
Scheme 1: Synthesis of 4. a) 1) EtLi, Et2O, −78 °C, 1 h; 2) (COOMe)2, Et2O, −78 °C, 2 h, 33%; b) FeCl3, MeNO2...
Alternatively, we prepared first 3,3’-thenil (2) by metalation of 3-bromothiophene and reaction with dimethyl oxalate [19]. Subsequent oxidative intramolecular thiophene-thiophene coupling [20] with FeCl3 yielded 3 as a dark red (nearly black) solid. Bromination of 3 with bromine in acetic acid/chloroform gave 4 in nearly quantitative yield [21,22]. The respective regio isomer 8 was synthesized starting from commercially available 3-bromothiophene 1 and boronic acid 5 in three steps (Scheme 2). By Suzuki–Miyaura coupling we obtained 3,3’-bithiophene in good yields. The red diketone 7 was prepared by two-fold acylation with oxalyl chloride [23,24]. Bromination of 7 with NBS failed, however, the reaction with bromine under similar conditions as described for 4 afforded 8 in quantitative yields. Both routes towards the brominated bithiophene-diketones gave high overall yields and can be scaled up easily.
![[1860-5397-6-135-i2]](/bjoc/content/inline/1860-5397-6-135-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 8. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 87%; b) oxalyl chloride, 1,2-dichloroethane, reflux, 85%; c) Br2, CHCl3, AcOH, reflux, 10 h, 97%.
Scheme 2: Synthesis of 8. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 87%; b) oxalyl chloride, 1,2-dich...
Condensation of the diketones 4 and 8 with diaminobenzenes 9a and 9b, containing methyl and hexyl side groups [25], in pure acetic acid at 50 °C gave the phenazines 10 and 11 as yellow solids in 50–93% yield (Scheme 3) [26]. The hexyl derivatives 10b and 11b showed a significantly higher solubility than the methyl analogues and could be more easily purified by column chromatography.
![[1860-5397-6-135-i3]](/bjoc/content/inline/1860-5397-6-135-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of phenazine isomers. a) AcOH, 50 °C, 2 h, 93% (10a), 53% (10b). b) AcOH, 50 °C, 2 h, 63% (11a), 50% (11b).
Scheme 3: Preparation of phenazine isomers. a) AcOH, 50 °C, 2 h, 93% (10a), 53% (10b). b) AcOH, 50 °C, 2 h, 6...
Nevertheless, both compounds can serve as starting materials for thiophene oligomers with increased donor ability. Thiophene- and bithiophene boronic acids or esters were coupled via Suzuki–Miyaura reactions forming orange-colored thiophene-tetramers 12a, 12b, 14 and 16 and red-colored hexamers 13, 15, 17a, 17b and 18, respectively (Scheme 4 and Scheme 5).
![[1860-5397-6-135-i4]](/bjoc/content/inline/1860-5397-6-135-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Suzuki–Miyaura reactions with 10a/10b. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 23% (12a), 36% (12b), 26% (13); b) Pd(PPh3)4, 1 M Cs2CO3, toluene, reflux, 19% (14), Pd(PPh3)4, 2 M Na2CO3, toluene, Aliquat 336, reflux, 21% (15).
Scheme 4: Suzuki–Miyaura reactions with 10a/10b. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 23% (12a),...
![[1860-5397-6-135-i5]](/bjoc/content/inline/1860-5397-6-135-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Suzuki–Miyaura reactions with 11a/11b. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 50% (16), 15% (17a), 19% (17b); b) Pd(PPh3)4, 2 M Na2CO3, toluene, Aliquat 336, reflux, 56% (18).
Scheme 5: Suzuki–Miyaura reactions with 11a/11b. a) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, reflux, 50% (16), ...
The low solubility of the methyl derivatives 12a, 16 and 17a made their purification tedious. However, this problem did not occur in the case of the hexyl substituted oligomers. Highly soluble compounds (20–50 mg/mL in chloroform or dichlorobenzene) were obtained by coupling of the hexyl substituted compounds 10b and 11b with alkyl substituted thiophene or bithiophene boronic acids (esters).
UV–Vis spectra
Figure 1a shows the UV–Vis absorption and emission spectra of 12a, 13, 14 and 15 in solution (CH2Cl2). In Figure 1b the spectra of the isomeric compounds 16, 17a and 18 are displayed [27]. All oligomers based on 10 exhibit strong absorption bands at ~250–350 nm and at ~350–450 nm with underlying vibronic structures. As expected, the longest wavelength absorption is for the hexathiophene derivatives shifted bathochromically in comparison to the tetrathiophene derivatives. The spectra of 16, 17a and 18 display two strong bands at ~300–400 nm and at ~400–500 nm, some of them show vibronic fine structures. The maxima of the emission spectra are shifted by ≈3500 to 4600 cm−1 with respect to the longest wavelength absorption maxima (Figure 1, Table 1).
![[1860-5397-6-135-1]](/bjoc/content/figures/1860-5397-6-135-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Absorption (solid lines) and emission (dotted lines) spectra of compounds 12a (blue), 13 (red), 14 (green) and 15 (black) in solution (CH2Cl2). b) Absorption (solid lines) and emission (dotted lines) spectra of compounds 16 (blue), 17a (red), 18 (black) in solution (CH2Cl2).
Figure 1: a) Absorption (solid lines) and emission (dotted lines) spectra of compounds 12a (blue), 13 (red), ...
Table 1: Spectral data of the synthesized oligomers. Absorption and emission spectra were measured in CH2Cl2 solutions. The zero–zero transition E0–0 values were estimated from the intersection of the absorption and emission spectra.
|
λabs.max
[nm] |
λabs.onset
[nm] |
λabs.onset
[eV] |
λem.max
[nm] |
Stokes shift
[cm−1] |
E0–0
[nm] |
E0–0
[eV] |
|
|---|---|---|---|---|---|---|---|
| 12a | 440 | 513 | 2.41 | 519 | 3459 | 463 | 2.68 |
| 14 | 462 | 550 | 2.25 | 583 | 4522 | 524 | 2.37 |
| 13 | 491 | 561 | 2.17 | 606 | 3865 | 531 | 2.33 |
| 15 | 495 | 577 | 2.13 | 622 | 4125 | 556 | 2.23 |
| 16 | 465 | 500 | 2.48 | 525 | 2457 | 493 | 2.51 |
| 17a | 478 | 530 | 2.34 | 585 | 3833 | 520 | 2.38 |
| 18 | 476 | 539 | 2.30 | 610 | 4615 | 531 | 2.33 |
At the onset of the absorption, the zero–zero transition and the longest wavelength absorption maxima follow the same (expected) trends, that is the more extended π-systems (13, 15, 17, 18) exhibit a smaller optical gap. Moreover, when compared with the optical spectra of the quater- and sexithiophene [28] (Table 2, 19 and 20), a clear red-shift could be observed for both oligomer series, regardless if they are based on 10 or 11, due to the intramolecular donor-acceptor character of the compounds.
Quantum chemical calculations
To rationalize the observed trends in the optical absorption spectra of the oligomers, DFT calculations using the ORCA program package [29] were carried out. The ground state geometries of quaterthiophene 19, the compounds 12a, 16, sexithiophene 20 and the compounds 13 and 17a were optimized with the B3LYP functional [30] with a TZVP basis set [31]. Different rotations of the thiophene rings were investigated each corresponding to a different local minimum-energy structure. The differences in the total energy of the ground states are within the typical DFT error range of 3 to 11 kJ/mol. It can therefore be expected that at room temperature all conformations are present. For simplicity we only present results for the most stable structure. For the treatment of excited states time-dependent density functional theory (TD-DFT) was applied. The COSMO model was used to simulate the solution environment of the molecules in CH2Cl2 [32,33].
The calculated spectra of the most stable conformers and the Sn–S0 difference electron densities of selected compounds are shown in Figure 2. In general, the calculated excitation energies are in reasonable agreement with the experimental results (see also Table 3). It has to be kept in mind that the calculated values correspond to vertical excitation energies between electronic states while the measured optical spectra include vibronic effects.
![[1860-5397-6-135-2]](/bjoc/content/figures/1860-5397-6-135-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Calculated absorption spectra of (a) 19, (b) 20, (c) 12a, (d) 13 with R=CH3, (e) 16 and (f) 17a. In every spectrum the ground state geometry and the difference densities of the excitations are shown. Blue (red) color indicates a decrease (increase) of electron density after transition from the ground state to the excited state. Absorption signals with large oscillator strengths are labeled with the main excitation inducing the transition to this state.
Figure 2: Calculated absorption spectra of (a) 19, (b) 20, (c) 12a, (d) 13 with R=CH3, (e) 16 and (f) 17a. In...
Table 3: Lowest vertical excitation energies obtained by B3LYP/TZVP TD-DFT calculations of the quaterthiophene 19 and derivatives as well as sexithiophene 20 and derivatives (gas phase and in CH2Cl2) in eV, and oscillator strengths fosc.
| State | 19 | 12a | 16 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gas | CH2Cl2 | fosc | Gas | CH2Cl2 | fosc | Gas | CH2Cl2 | fosc | |
| S1 | 3.14 | 3.13 | 1.39 | 2.37 | 2.11 | 0.19 | 2.69 | 2.56 | 0.26 |
| S2 | 4.80 | 4.78 | 0.13 | 3.18 | 3.18 | 1.02 | 3.53 | 3.49 | 1.17 |
| State | 20 | 13 (R=CH3) | 17a | ||||||
| Gas | CH2Cl2 | fosc | Gas | CH2Cl2 | fosc | Gas | CH2Cl2 | fosc | |
| S1 | 2.64 | 2.63 | 2.27 | 2.21 | 1.97 | 0.45 | 2.43 | 2.21 | 0.08 |
| S2 | 3.74 | 3.73 | 0.35 | 3.18 | 3.15 | 1.46 | 2.46 | 2.24 | 0.30 |
The dominating signals in the spectra of quaterthiophene 19 and sexithiophene 20 correspond to the HOMO-LUMO excitation. While the calculated S1 excitation energy of 19 differs by about 0.4 eV from the experimental result, the excitation energy of 20 is within the typical error range of B3LYP (ΔEexp-calc = 0.1 eV) [34]. Both compounds have a similar spectrum, but the transitions of 20 are red shifted due to its larger delocalized π-system, which lowers the HOMO-LUMO gap as discussed above.
The difference density of 12a (Figure 2c) shows that the S0-S1 excitation leads to an intramolecular charge transfer (CT) from the thiophenes to the phenazine part of the molecule. These calculations verify our initial assumption that the electron rich thiophene units act as electron donors and the electron poor phenazine unit as an electron acceptor in this intramolecular CT-complex. This CT state can also be observed in 16 (Figure 2e), but here the calculated difference between the S1 energies of 12a and 16 (0.27 eV) is larger than the measured value (0.07 eV). Nevertheless, the computed excitation energies show a similar trend as the observed maxima of the absorption spectra.
The increased length of the thiophene chains in 13 compared to 12a lowers the excitation energies of the CT states by about 0.2 eV due to an extended electron delocalization, as it is also experimentally observed. The same holds for the isomeric series. The calculated and observed optical gap in 17a is considerably smaller than in 16. It is worth noting that in both hexathiophene derivatives the charge reorganization upon excitation includes also the outermost thiophene rings, indicating that an extension of the chain would further reduce the optical gap. In addition, 17a has a nearly degenerate S2 state, which also has CT properties. This HOMO-1 to LUMO excitation is visible in 17a but not in 13 since the orbital energy and shape of HOMO and HOMO-1 in 17a are nearly the same (ΔEHOMO,HOMO-1 =0.02 eV), while in 13 the energy difference between HOMO and HOMO-1 is larger than 0.6 eV [35].
STM study
All synthesized thiophene oligomers 12–18 were investigated concerning their ability to form self-assembled monolayers (SAMs) at the HOPG/TCB interface (highly oriented pyrolytic graphite/1,2,4-trichlorobenzene). In situ STM was applied under typical conditions with typical tunneling parameters normally allowing the visualization of SAMs – if formed [36]. However, among all the substances only 12b (10−5 M in TCB) assembles into crystalline monolayers on HOPG (Figure 3a). Bright and dark colors originate from locally high and low tunneling currents, resulting from unsaturated (backbone) and saturated (alkyl side chain) hydrocarbon segments, respectively [37]. A unit cell with a = 2.5 ± 0.1 nm, b = 2.3 ± 0.1 nm, γ = 90 ± 2° could be determined [38]. A proposed molecular model of the adsorbate geometry is shown in Figure 3b.
![[1860-5397-6-135-3]](/bjoc/content/figures/1860-5397-6-135-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Self-assembled monolayer of 12b on HOPG. a) STM image (VS = −0.8 V, It = 80 pA, image size 25.0 × 25.0 nm2); b) structure model [39].
Figure 3: Self-assembled monolayer of 12b on HOPG. a) STM image (VS = −0.8 V, It = 80 pA, image size 25.0 × 2...
The banana-shaped bright features in Figure 3a are attributed to the backbones of 12b, assembling in densely packed double rows. However, short alkyl chains are commonly not visible under the applied conditions. Nevertheless we assume they are oriented along the main crystallographic direction of the HOPG substrate.
Ordered adlayers were observed only for 12b, but not for 12a. Moreover, additional terminal alkyl chains terminating the end-capping thiophene substituents of 14, 15 and 18 appear to hinder the formation of a self-assembled monolayer, most probably due to sterical hindrance of the molecules to cover the surface densely.
Conclusion
We have developed a simple method for preparing phenazine-thiophene oligomers. These compounds show interesting optical characteristics, which can be tuned by attachment of different thiophene substituents. Currently we investigate the electrooptical properties of these compounds and similar derivatives. In addition, for one of the compounds a SAM on HOPG could be observed.
Supporting Information
Experimental procedures and 1H and 13C NMR spectra and MS data for all new compounds are supplied, including X-ray data and a cif-file for 4 and 7.
| Supporting Information File 1: Syntheses and properties of thienyl-substituted dithienophenazines, experimental procedures, and characterization. | ||
| Format: PDF | Size: 957.7 KB | Download |
| Supporting Information File 2: Syntheses and properties of thienyl-substituted dithienophenazines – cif files. | ||
| Format: CIF | Size: 30.4 KB | Download |
References
-
Fichou, D., Ed. Handbook of Oligo- and Polythiophenes; Wiley-VCH: Weinheim, Germany, 1999.
Return to citation in text: [1] -
Mishra, A.; Ma, C.-Q.; Bäuerle, P. Chem. Rev. 2009, 109, 1141–1276. doi:10.1021/cr8004229
Return to citation in text: [1] -
Perepichka, I. F.; Perepichka, D. F.; Meng, H.; Wudl, F. Adv. Mater. 2005, 17, 2281–2305. doi:10.1002/adma.200500461
Return to citation in text: [1] -
Müllen, K.; Wegner, G., Eds. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998.
Return to citation in text: [1] -
Zhu, Z.; Waller, D.; Brabec, C. J. New Construction of Low-Bandgap Conducting Polymers. In Organic Photovoltaics; Brabec, C.; Dyakonov, V.; Scherf, U., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp 129–152.
Return to citation in text: [1] -
Chen, J.; Cao, Y. Acc. Chem. Res. 2009, 42, 1709–1718. doi:10.1021/ar900061z
Return to citation in text: [1] -
Thompson, B. C.; Fréchet, J. M. J. Angew. Chem. 2008, 120, 62–82. doi:10.1002/ange.200702506
Return to citation in text: [1] -
Bundgaard, E.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2007, 91, 954–985. doi:10.1016/j.solmat.2007.01.015
Return to citation in text: [1] -
Yan, Q.; Zhou, Y.; Ni, B.-B.; Ma, Y.; Wang, J.; Pei, J.; Cao, Y. J. Org. Chem. 2008, 73, 5328–5339. doi:10.1021/jo800606b
Return to citation in text: [1] -
Tovar, J. D.; Swager, T. M. Adv. Mater. 2001, 13, 1775–1780. doi:10.1002/1521-4095(200112)13:23<1775::AID-ADMA1775>3.0.CO;2-N
Return to citation in text: [1] -
Velusamy, M.; Huang, J.-H.; Hsu, Y.-C.; Chou, H.-H.; Ho, K.-C.; Wu, P.-L.; Chang, W.-H.; Lin, J. T.; Chu, C.-W. Org. Lett. 2009, 11, 4898–4901. doi:10.1021/ol9019953
Return to citation in text: [1] -
Rieger, R.; Beckmann, D.; Pisula, W.; Steffen, W.; Kastler, M.; Müllen, K. Adv. Mater. 2010, 22, 83–86. doi:10.1002/adma.200901286
Return to citation in text: [1] -
Xiao, S.; Zhou, H.; You, W. Macromolecules 2008, 41, 5688–5696. doi:10.1021/ma800776q
Return to citation in text: [1] -
Pan, G.-B.; Cheng, X.-H.; Höger, S.; Freyland, W. J. Am. Chem. Soc. 2006, 128, 4218–4219. doi:10.1021/ja060469f
Return to citation in text: [1] -
Mena-Osteritz, E.; Bäuerle, P. Adv. Mater. 2006, 18, 447–451. doi:10.1002/adma.200501575
Return to citation in text: [1] -
Chen, T.; Pan, G.-B.; Wettach, H.; Fritzsche, M.; Höger, S.; Wan, L.-J.; Yang, H.-B.; Northrop, B. H.; Stang, P. J. J. Am. Chem. Soc. 2010, 132, 1328–1333. doi:10.1021/ja907220f
Return to citation in text: [1] -
Piot, L.; Silly, F.; Tortech, L.; Nicolas, Y.; Blanchard, P.; Roncalli, J.; Fichou, D. J. Am. Chem. Soc. 2009, 131, 12864–12865. doi:10.1021/ja902621t
Return to citation in text: [1] -
Wynberg, H.; Sinnige, H. J. M. Rec. Trav. Chim. 1969, 88, 1244–1245. doi:10.1002/recl.19690881011
Return to citation in text: [1] -
Nyberg, K. Acta Chem. Scand. 1969, 23, 1087–1089. doi:10.3891/acta.chem.scand.23-1087
Return to citation in text: [1] -
Tovar, J. D.; Rose, A.; Swager, T. M. J. Am. Chem. Soc. 2002, 124, 7762–7769. doi:10.1021/ja0262636
Return to citation in text: [1] -
Khor, E.; Siu, C. N.; Hwee, C. L.; Chai, S. Heterocycles 1991, 32, 1805–1812. doi:10.3987/COM-91-5812
This reference gives the reaction conditions.
Return to citation in text: [1] -
The single crystal X-ray analysis data for 4 are given in the Supporting Information (File 1).
Return to citation in text: [1] -
Phillips, K. E. S.; Katz, T. J.; Jockusch, S.; Lovinger, A. J.; Turro, N. J. J. Am. Chem. Soc. 2001, 123, 11899–11907. doi:10.1021/ja011706b
Return to citation in text: [1] -
The single crystal X-ray analysis data for 7 are given in the Supporting Information (File 1).
Return to citation in text: [1] -
Zhang, F.; Bai, S.; Yap, G. P. A.; Fox, J. M. J. Am. Chem. Soc. 2005, 127, 10590–10599. doi:10.1021/ja050886c
Return to citation in text: [1] -
Gadisa, A.; Mammo, W.; Andersson, M.; Admassie, S.; Zhang, F.; Andersson, M. R.; Inganäs, O. Adv. Funct. Mat. 2007, 17, 3836–3842. doi:10.1002/adfm.200700441
Return to citation in text: [1] -
The spectra of compounds 12b und 17b are nearly identical to the spectra of 12a and 17a and are not shown.
Return to citation in text: [1] -
19 and 20 are displayed in the Supporting Information (File 1).
Return to citation in text: [1] -
ORCA – an ab initio, Density Functional and Semiempirical program package, Version 2.7; University of Bonn, 2009.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Schäfer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829–5835. doi:10.1063/1.467146
Return to citation in text: [1] -
Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J. C. W. J. Phys. Chem. A 1998, 102, 5074–5085. doi:10.1021/jp980017s
Return to citation in text: [1] -
Sinnecker, S.; Rajendran, A.; Klamt, A.; Diedenhofen, M.; Neese, F. J. Phys. Chem. A 2006, 110, 2235–2245. doi:10.1021/jp056016z
Return to citation in text: [1] -
Dreuw, A.; Head-Gordon, M. Chem. Rev. 2005, 105, 4009–4037. doi:10.1021/cr0505627
Return to citation in text: [1] -
The calculated solvent shift is negligible for the parent compounds 19 and 20 (Table 2 and Table 3). Due to their CT character, the bathochromic shift is more pronounced for the S1 states of the substituted compounds 12a, 16, 13 and 17a.
Return to citation in text: [1] -
A drop of the respective solution (approx. 1 mg/ml) in TCB (1,2,4-trichlorobenzene) was applied to a piece of freshly cleaved HOPG, and STM was performed at voltages between −(0.4–1.2V) and currents between 2–100 pA.
Return to citation in text: [1] -
The tunneling efficiency depends on the energetic difference of the tip level and the levels of the frontier orbitals of the respective molecular segments. For theoretical descriptions of the contrast mechanism in STM see i.e. [40,41].
Return to citation in text: [1] -
All acquired STM images were in situ calibrated by (immediately and) subsequently reducing the tip bias to −50 mV, and acquiring the atomically resolved lattice of the HOPG substrate (without changing the position), using the latter as a calibration grid for the images of the adlayer. Respective data handling is done with the SPIP software package (Image Metrology, Inc.).
Return to citation in text: [1] -
The resolution of STM is not sufficient to distinguish between cisoid and transoid conformers of the oligothiophene. However, in the molecular model we displayed the adsorbed molecule in transoid conformation in accordance with the TD-DFT calculation.
Return to citation in text: [1] -
Lazzaroni, R.; Calderone, A.; Brédas, J. L.; Rabe, J. P. J. Chem. Phys. 1997, 107, 99–105. doi:10.1063/1.474356
Return to citation in text: [1] -
Sautet, P. Chem. Rev. 1997, 97, 1097–1116. doi:10.1021/cr9600823
Return to citation in text: [1]
| 1. | Fichou, D., Ed. Handbook of Oligo- and Polythiophenes; Wiley-VCH: Weinheim, Germany, 1999. |
| 2. | Mishra, A.; Ma, C.-Q.; Bäuerle, P. Chem. Rev. 2009, 109, 1141–1276. doi:10.1021/cr8004229 |
| 3. | Perepichka, I. F.; Perepichka, D. F.; Meng, H.; Wudl, F. Adv. Mater. 2005, 17, 2281–2305. doi:10.1002/adma.200500461 |
| 14. | Pan, G.-B.; Cheng, X.-H.; Höger, S.; Freyland, W. J. Am. Chem. Soc. 2006, 128, 4218–4219. doi:10.1021/ja060469f |
| 15. | Mena-Osteritz, E.; Bäuerle, P. Adv. Mater. 2006, 18, 447–451. doi:10.1002/adma.200501575 |
| 16. | Chen, T.; Pan, G.-B.; Wettach, H.; Fritzsche, M.; Höger, S.; Wan, L.-J.; Yang, H.-B.; Northrop, B. H.; Stang, P. J. J. Am. Chem. Soc. 2010, 132, 1328–1333. doi:10.1021/ja907220f |
| 17. | Piot, L.; Silly, F.; Tortech, L.; Nicolas, Y.; Blanchard, P.; Roncalli, J.; Fichou, D. J. Am. Chem. Soc. 2009, 131, 12864–12865. doi:10.1021/ja902621t |
| 29. | ORCA – an ab initio, Density Functional and Semiempirical program package, Version 2.7; University of Bonn, 2009. |
| 9. | Yan, Q.; Zhou, Y.; Ni, B.-B.; Ma, Y.; Wang, J.; Pei, J.; Cao, Y. J. Org. Chem. 2008, 73, 5328–5339. doi:10.1021/jo800606b |
| 10. | Tovar, J. D.; Swager, T. M. Adv. Mater. 2001, 13, 1775–1780. doi:10.1002/1521-4095(200112)13:23<1775::AID-ADMA1775>3.0.CO;2-N |
| 11. | Velusamy, M.; Huang, J.-H.; Hsu, Y.-C.; Chou, H.-H.; Ho, K.-C.; Wu, P.-L.; Chang, W.-H.; Lin, J. T.; Chu, C.-W. Org. Lett. 2009, 11, 4898–4901. doi:10.1021/ol9019953 |
| 12. | Rieger, R.; Beckmann, D.; Pisula, W.; Steffen, W.; Kastler, M.; Müllen, K. Adv. Mater. 2010, 22, 83–86. doi:10.1002/adma.200901286 |
| 13. | Xiao, S.; Zhou, H.; You, W. Macromolecules 2008, 41, 5688–5696. doi:10.1021/ma800776q |
| 5. | Zhu, Z.; Waller, D.; Brabec, C. J. New Construction of Low-Bandgap Conducting Polymers. In Organic Photovoltaics; Brabec, C.; Dyakonov, V.; Scherf, U., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp 129–152. |
| 6. | Chen, J.; Cao, Y. Acc. Chem. Res. 2009, 42, 1709–1718. doi:10.1021/ar900061z |
| 7. | Thompson, B. C.; Fréchet, J. M. J. Angew. Chem. 2008, 120, 62–82. doi:10.1002/ange.200702506 |
| 8. | Bundgaard, E.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2007, 91, 954–985. doi:10.1016/j.solmat.2007.01.015 |
| 27. | The spectra of compounds 12b und 17b are nearly identical to the spectra of 12a and 17a and are not shown. |
| 4. | Müllen, K.; Wegner, G., Eds. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998. |
| 21. |
Khor, E.; Siu, C. N.; Hwee, C. L.; Chai, S. Heterocycles 1991, 32, 1805–1812. doi:10.3987/COM-91-5812
This reference gives the reaction conditions. |
| 22. | The single crystal X-ray analysis data for 4 are given in the Supporting Information (File 1). |
| 25. | Zhang, F.; Bai, S.; Yap, G. P. A.; Fox, J. M. J. Am. Chem. Soc. 2005, 127, 10590–10599. doi:10.1021/ja050886c |
| 20. | Tovar, J. D.; Rose, A.; Swager, T. M. J. Am. Chem. Soc. 2002, 124, 7762–7769. doi:10.1021/ja0262636 |
| 26. | Gadisa, A.; Mammo, W.; Andersson, M.; Admassie, S.; Zhang, F.; Andersson, M. R.; Inganäs, O. Adv. Funct. Mat. 2007, 17, 3836–3842. doi:10.1002/adfm.200700441 |
| 19. | Nyberg, K. Acta Chem. Scand. 1969, 23, 1087–1089. doi:10.3891/acta.chem.scand.23-1087 |
| 18. | Wynberg, H.; Sinnige, H. J. M. Rec. Trav. Chim. 1969, 88, 1244–1245. doi:10.1002/recl.19690881011 |
| 23. | Phillips, K. E. S.; Katz, T. J.; Jockusch, S.; Lovinger, A. J.; Turro, N. J. J. Am. Chem. Soc. 2001, 123, 11899–11907. doi:10.1021/ja011706b |
| 24. | The single crystal X-ray analysis data for 7 are given in the Supporting Information (File 1). |
| 34. | Dreuw, A.; Head-Gordon, M. Chem. Rev. 2005, 105, 4009–4037. doi:10.1021/cr0505627 |
| 31. | Schäfer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829–5835. doi:10.1063/1.467146 |
| 32. | Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J. C. W. J. Phys. Chem. A 1998, 102, 5074–5085. doi:10.1021/jp980017s |
| 33. | Sinnecker, S.; Rajendran, A.; Klamt, A.; Diedenhofen, M.; Neese, F. J. Phys. Chem. A 2006, 110, 2235–2245. doi:10.1021/jp056016z |
| 39. | The resolution of STM is not sufficient to distinguish between cisoid and transoid conformers of the oligothiophene. However, in the molecular model we displayed the adsorbed molecule in transoid conformation in accordance with the TD-DFT calculation. |
| 40. | Lazzaroni, R.; Calderone, A.; Brédas, J. L.; Rabe, J. P. J. Chem. Phys. 1997, 107, 99–105. doi:10.1063/1.474356 |
| 41. | Sautet, P. Chem. Rev. 1997, 97, 1097–1116. doi:10.1021/cr9600823 |
| 37. | The tunneling efficiency depends on the energetic difference of the tip level and the levels of the frontier orbitals of the respective molecular segments. For theoretical descriptions of the contrast mechanism in STM see i.e. [40,41]. |
| 38. | All acquired STM images were in situ calibrated by (immediately and) subsequently reducing the tip bias to −50 mV, and acquiring the atomically resolved lattice of the HOPG substrate (without changing the position), using the latter as a calibration grid for the images of the adlayer. Respective data handling is done with the SPIP software package (Image Metrology, Inc.). |
| 35. | The calculated solvent shift is negligible for the parent compounds 19 and 20 (Table 2 and Table 3). Due to their CT character, the bathochromic shift is more pronounced for the S1 states of the substituted compounds 12a, 16, 13 and 17a. |
| 36. | A drop of the respective solution (approx. 1 mg/ml) in TCB (1,2,4-trichlorobenzene) was applied to a piece of freshly cleaved HOPG, and STM was performed at voltages between −(0.4–1.2V) and currents between 2–100 pA. |
© 2010 Meyer et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)