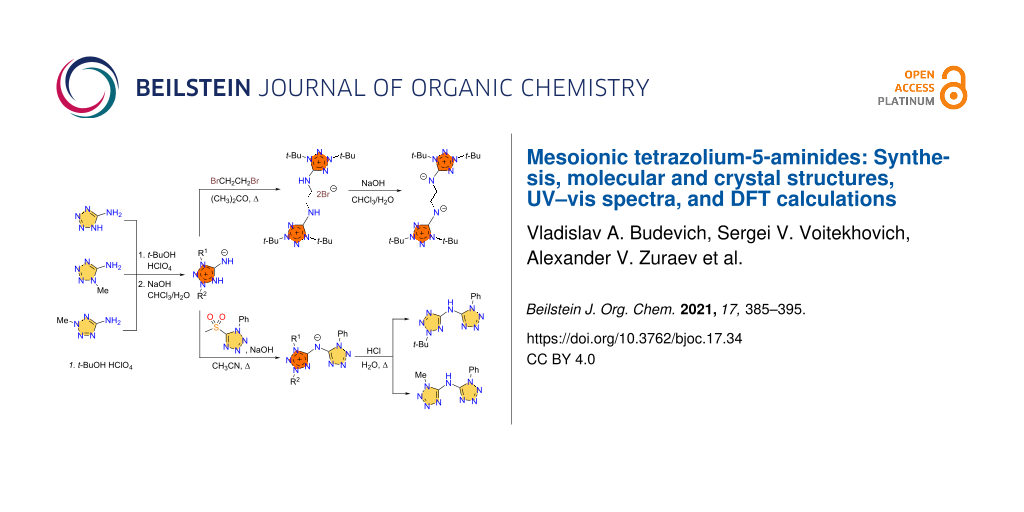
Abstract
Tetrazolium-5-aminides have been prepared by the tert-butylation of 5-aminotetrazole and its N-methyl derivatives by the t-BuOH/HClO4 system followed by the treatment of the tetrazolium salts by alkali. The mesoionic compounds have been found to show a higher reactivity of the exocyclic N atom in comparison with 5-aminotetrazoles. The compounds reacted with 1,2-dibromoethane and 5-(methylsulfonyl)-1-phenyl-1H-tetrazole with substitution of bromine and methylsulfonyl groups giving the corresponding tetrazolium salts or conjugate aminides. The obtained mesoionic tetrazoles have been characterized by elemental analysis, FTIR, NMR, and UV–vis spectroscopy, TGA/DSC analysis and for 1,3-di-tert-butyltetrazolium-5-aminide, its N,N’-ethylene-bridged bis-derivative and (1,3-di-tert-butyl-1H-tetrazol-3-ium-5-yl)(1-phenyl-1H-tetrazol-5-yl)amide by single crystal X-ray analysis. The structural and spectral features of the tetrazolium-5-aminides are discussed by using quantum-chemical calculations.

Graphical Abstract
Introduction
5-Aminotetrazoles are one of the most available and valuable tetrazole derivatives. So, due to the thermal stability and high nitrogen content the parent 5-aminotetrazole (1, Figure 1) is of practical interest as a gas-generator and blowing agent [1,2]. Moreover, it is a useful building block in organic synthesis, including various multicomponent reactions opening the way to diverse fused heterocycles [3]. Salts with anionic tetrazole, i.e., aminotetrazolates 2, and cationic ones, i.e., aminotetrazolium salts 3, are attractive as environmentally friendly pyrotechnics [4], insensitive high-energy materials [5-7], and promising energetic ionic liquids [8-10].
![[1860-5397-17-34-1]](/bjoc/content/figures/1860-5397-17-34-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The most interesting and less examined 5-aminotetrazole derivatives are tetrazolium-5-aminides, which can be depicted as 4 by analogy with other mesoionic tetrazoles [11,12]. The first representatives of tetrazole-5-aminides were reported in the 1950s [13,14], whereas next publications were appeared only five decades later, being devoted to the synthesis and photochemistry of 1,3-diaryltetrazolium-5-aminides [15-21]. NMR studies of a few tetrazole-5-aminides were carried out as a part of studies of mesoionic compounds [22-24]. Earlier, we reported the facile preparation of 1,3-di-tert-butyltetrazolium-5-aminide [25], which was later used for the synthesis of the first tetrazolium halocuprate [26] and also shown to react with bromine-containing alkylation reagents [27]. Very recently, this aminide was found to be a suitable ligand for manganese complexes [28] and used as the agent for the preparation of salts with high energy density [29]. Also, it is worth noting that today only a few examples of mesoionic tetrazole aminide X-ray structures are known [17,28].
Thus, the information on tetrazole-5-aminides is very limited and fragmentary. In the present work, we try to fill this gap by carrying out experimental (synthesis, X-ray, UV–vis) and theoretical density functional theory (DFT) studies of some selected 1,3-dialkyltetrazolium-5-aminides.
Results and Discussion
Synthesis and chemical transformations
Tetrazolium-5-aminides can be prepared by three main approaches: a) by the deprotonation of 5-aminotetrazolium salts, b) by the photochemical transformation of 5-azidotetrazolium salts, and c) by the functionalization of other aminides. The first route is preferable due to the synthetic availability of aminotetrazolium salts [25]. Therefore, here we synthesized 1,3-disubstituted 5-aminotetrazolium perchlorates 7 by the quaternization of commercial 5-aminotetrazole (1) and its N-methylated derivatives 5 and 6 as shown in Scheme 1. The quaternization proceeded regioselectively using the t-BuOH/HClO4 system [25]. Further, the salts 7 were treated with sodium hydroxide in the biphasic system water/chloroform giving aminides 8, which were extracted from the reaction mixtures by chloroform.
![[1860-5397-17-34-i1]](/bjoc/content/inline/1860-5397-17-34-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of tetrazolium-5-aminides.
Scheme 1: Synthesis of tetrazolium-5-aminides.
The obtained aminides 8 are yellow solids and soluble in various organic solvents, such as alcohols, chloroform, dichloromethane, hexane, acetonitrile, toluene, and THF. They are also soluble in water. Remarkably, the solutions in organic solutions are yellow colored, whereas aqueous solutions are colorless. The UV–vis spectra of 8a were found to show solvatochromism, that is discussed in more detail in the theoretical section. The 13C NMR shift of the C5 endocyclic atom for aminides 8 is observed at 162.1–163.1 ppm in DMSO-d6. For the parent 5-aminotetrazolium salts, this chemical shift is found at 156.5–158.3 ppm in the same solvent. The TGA/DSC data show thermal stability of the tetrazolium-5-aminides (see Supporting Information File 1 for more details).
Some nucleophilic displacement reactions were carried out in order to show the higher reactivity of the aminides in comparison to the parent aminotetrazoles. The high nucleophilicity of the imine group in tetrazolium-5-aminide allows to displace halo- and methylsulfonyl groups, whereas 5-aminotetrazoles do not react under analogous conditions. So, we prepared the bistetrazolium salt 9 by the alkylation of aminide 8a with 1,2-dibromoethane (Scheme 2). The obtained salt 9 was subjected to deprotonation to give the bistetrazolium-5-aminide 10. The reactions of aminides 8a and 8b with 5-methylsulfonyl-1-phenyltetrazole in the presence of sodium hydroxide in boiling acetonitrile yielded compounds 11a and 11b, respectively.
![[1860-5397-17-34-i2]](/bjoc/content/inline/1860-5397-17-34-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: N-Functionalizations of 1,3-disubstituted tetrazolium-5-aminides 8a,b.
Scheme 2: N-Functionalizations of 1,3-disubstituted tetrazolium-5-aminides 8a,b.
Taking into account the possibility of removing the tert-butyl group in tetrazolium salts under acidic conditions [30], N-tert-butyltetrazolium-5-aminides are of special interest as agents for the introduction of tetrazol-5-ylamino groups into various substrates having suitable leaving groups. Therefore, we carried out the de-tert-butylation of compounds 11a and 11b under the action of hydrochloric acid. Indeed, the de-tert-butylation was found to be regioselective leading to the 2-tert-butyltetrazole (12a) and 1-methyltetrazole (12b) derivatives, respectively (Scheme 2). The selectivity of the reaction was confirmed by a 13C NMR shift comparison for the C5 endocyclic atoms of tetrazoles 12a and 12b: 161.1 and 151.9 ppm for compound 12a and 153.2 and 151.1 ppm for compound 12b. It is known that the 13C NMR shifts for the endocyclic C5 atom in 2,5-disubstituted tetrazoles is located downfield (162–167 ppm and 151.9 ppm for 2-methyl-5H-tetrazole) from the corresponding signal of the 1,5-regioisomers (152–156 and 143.4 ppm for 1-methyl-5H-tetrazole) [31]. The de-tert-butylation selectivity observed for compound 11b can be explained by the higher stability of the t-Bu cation versus the Me cation.
Crystal structures
The mesoionic compounds 8a, 10, 11a, and salt 9 were characterized by single crystal X-ray analysis. For all compounds, data collection was performed at a temperature of 100 K and the main crystal data and structure refinement details are given in Table 1.
Table 1: Single crystal X-ray data and structure refinement details for compounds 8a, 10, 11a, and 9.
| 8a | 10 | 11a | 9 | |
| empirical formula | C9H19N5 | C20H40N10 | C16H23N9 | C20H42Br2N10 |
| formula weight | 197.29 | 420.62 | 341.43 | 582.45 |
| temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| crystal system | monoclinic | monoclinic | monoclinic | trigonal |
| space group | P21/c | P21/c | P21/n |
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-34-i5.svg?max-width=637&scale=1.0)
|
| a (Å) | 5.94661(7) | 8.23030(10) | 9.04560(10) | 36.8562(4) |
| b (Å) | 16.2954(2) | 10.4082(2) | 9.99730(10) | 36.8562(4) |
| c (Å) | 12.04281(15) | 14.7565(2) | 20.0685(3) | 12.05390(10) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 100.2696(6) | 94.5711(7) | 100.7743(4) | 90 |
| γ (°) | 90 | 90 | 90 | 120 |
| V (Å3) | 1148.28(2) | 1260.06(3) | 1782.83(4) | 14180.1(3) |
| Z | 4 | 2 | 4 | 18 |
| dc (g cm–3) | 1.141 | 1.109 | 1.272 | 1.228 |
| μ (mm–1) | 0.074 | 0.072 | 0.084 | 2.596 |
| crystal size (mm) | 0.56 × 0.31 × 0.22 | 0.58 × 0.50 × 0.49 | 0.40 × 0.28 × 0.18 | 0.50 × 0.10 × 0.09 |
| refl. collected | 32431 | 29074 | 54434 | 108400 |
| refl. independ. | 4411 | 3863 | 8700 | 9633 |
| restraints | 0 | 0 | 0 | 0 |
| parameters | 203 | 196 | 295 | 301 |
| GOOF on F2 | 1.029 | 1.072 | 1.063 | 1.039 |
| R1/wR2 [I> 2σ(I)] | 0.0335/0.0919 | 0.0371/0.0982 | 0.0384/0.1027 | 0.0383/0.0796 |
| R1/wR2 [all data] | 0.0361/0.0953 | 0.0392/0.0998 | 0.0461/0.1095 | 0.0639/0.0871 |
| # CCDC | 2035296 | 2035297 | 2035298 | 2035299 |
The mesoionic compounds 8a, 10, and 11a all are monoclinic, with the space groups P21/c for 8a and 10, and P21/n for 11a, respectively. The asymmetric units of compounds 8a and 11a contain one molecule, whereas the unit of 10 includes a half molecule. In contrast to 8a and 11a, the molecule of 10 shows Ci symmetry, with the inversion center lying in the middle of the bond C10–C10a [symmetry code: (a) –x + 2, −y, −z + 1]. The structures of the molecules are shown in Figure 2.
![[1860-5397-17-34-2]](/bjoc/content/figures/1860-5397-17-34-2.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 2: Molecules of compounds 8a, 10, 11a, and the bistetrazolium cation 9, with displacement ellipsoids drawn at the 50% probability level. The hydrogen atoms are shown as spheres of arbitrary radii. The atom numbering is done for the asymmetric unit.
Figure 2: Molecules of compounds 8a, 10, 11a, and the bistetrazolium cation 9, with displacement ellipsoids d...
The bond lengths in the tetrazole cycles and exocyclic C–N bonds in compounds 8a, 10, and 11a are given in Table 2. In these compounds, the shortest bonds are the endocyclic N2–N3 and the exocyclic C5–N5 bonds, being close to double bonds in lengths. However, note that the C5–N5 bond in compound 11a is somewhat longer compared to those in 8a and 10. This fact can be attributed to some electron density shift in 11a from this bond to the neighboring N5–C15 bond, which is exocyclic in the other tetrazole ring N11/C15 and shows the length of 1.3497(7) Å. The longest endocyclic bonds are N1–C5 and N4–C5, lying in the ranges 1.3892(7)–1.4101(9) and 1.3551(7)–1.3784(9) Å, respectively. The remaining N1–N2 and N3–N4 bonds show close lengths, ranging from 1.3333(7) to 1.3419(6) Å.
Table 2: The lengths of the tetrazole ring and exocyclic C–N bonds (Å) in compounds 8a, 10, and 11a, and in the 1,3-di-tert-butyltetrazolium-5-aminide ligand in manganese complexes [29].
| entry | bond | 8a | 10 | 11a | reference [29] |
| 1 | N1–C5 | 1.4034(6) | 1.4101(9) | 1.3892(7) | 1.386(3), 1.390(3) |
| 2 | N1–N2 | 1.3419(6) | 1.3396(8) | 1.3333(7) | 1.352(3), 1.346(3) |
| 3 | N2–N3 | 1.2937(6) | 1.2944(8) | 1.2935(7) | 1.295(3), 1.291(3) |
| 4 | N3–N4 | 1.3350(6) | 1.3397(8) | 1.3411(7) | 1.331(3), 1.337(3) |
| 5 | N4–C5 | 1.3784(7) | 1.3842(9) | 1.3551(7) | 1.355(3), 1.358(3) |
| 6 | C5–N5 | 1.2986(7) | 1.2869(9) | 1.3241(7) | 1.316(3), 1.315(3) |
In Table 2, we also included the bond lengths of the 1,3-di-tert-butyltetrazolium-5-aminide ligand in a manganese complex [29], being the only structurally characterized complex with a neutral 1,3-dialkyltetrazolium-5-aminide. Therefore, it is of interest to compare the structural data for this compound with those obtained for compound 8a to find the influence of the complexation on the ligand structure. As can be seen, the bond lengths of the free 1,3-di-tert-butyltetrazolium-5-aminide 8a and the ligand in the manganese complex are rather close. Nevertheless, the following structural differences attract attention. In the complex, the endocyclic N1–C5 and N4–C5 bonds are shorter, but the exocyclic C5–N5 bond is longer compared to aminide 8a. One can expect that these structural differences are due to the complexation.
It should be noted that the bond lengths of the tetrazole ring N11/C15 in compound 11a are usual for 1- and 1,5-substituted tetrazoles. The compounds 8a, 10, and 11a show no hydrogen bonds in their crystal structures and only van der Waals interactions take place between the molecules.
The bistetrazolium salt 9 (bromide salt of mesoionic compound 10) crystallizes in the trigonal space group ![[Graphic 3]](/bjoc/content/inline/1860-5397-17-34-i6.svg?max-width=637&scale=1.18182) , with 18 formula units in the unit cell. The asymmetric unit includes one cation, shown in Figure 2, and two bromide anions. The structure of the cation is close to C2 symmetry, with an rms deviation of its non-hydrogen atoms from ideal positions of 0.1320 Å. In the salt 9, the lengths of the tetrazole ring bonds and the exocyclic C–N bond are given in Table 3 together with the data for 1,3-di-alkyltetrazolium-5-aminide salts as described in the literature [25,26,28,32-35]. First of all, it should be mentioned that the formation of the salt from the corresponding mesoionic compound followed by protonation of the endocyclic N atom in all cases presented in Table 3. As can be seen, a good agreement of the bond lengths is observed for each of the six bonds presented in Table 3, and the methyl- and tert-butyl derivatives show no differences. On the other hand, a comparison of the data in Table 2 and Table 3 reveals the following structural differences of the salts and the mesoionic compounds: a) the bonds 1 and 5 in the salts are shorter in comparison with the mesoionic compounds and b) the bonds 6 (exocyclic C–N) are somewhat longer in the salts. Hence, when the salt is formed the same trends in structural changes in the mesoions are observed as under their complexation.
, with 18 formula units in the unit cell. The asymmetric unit includes one cation, shown in Figure 2, and two bromide anions. The structure of the cation is close to C2 symmetry, with an rms deviation of its non-hydrogen atoms from ideal positions of 0.1320 Å. In the salt 9, the lengths of the tetrazole ring bonds and the exocyclic C–N bond are given in Table 3 together with the data for 1,3-di-alkyltetrazolium-5-aminide salts as described in the literature [25,26,28,32-35]. First of all, it should be mentioned that the formation of the salt from the corresponding mesoionic compound followed by protonation of the endocyclic N atom in all cases presented in Table 3. As can be seen, a good agreement of the bond lengths is observed for each of the six bonds presented in Table 3, and the methyl- and tert-butyl derivatives show no differences. On the other hand, a comparison of the data in Table 2 and Table 3 reveals the following structural differences of the salts and the mesoionic compounds: a) the bonds 1 and 5 in the salts are shorter in comparison with the mesoionic compounds and b) the bonds 6 (exocyclic C–N) are somewhat longer in the salts. Hence, when the salt is formed the same trends in structural changes in the mesoions are observed as under their complexation.
Table 3: The bond lengths of the tetrazole ring and exocyclic C–N bonds (Å) in salt 9 and the corresponding literature data for 1,3-di-alkyltetrazolium-5-aminide saltsa,b.
| bond numbering |
bond
# |
salt 9 |
R = Me
[32-35] |
R = t-Bu
[25,26,28] |
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-34-i4.svg?max-width=637&scale=1.0)
|
1 | 1.366(3), 1.359(3) | 1.354–1.360 | 1.365–1.370 |
| 2 | 1.344(2), 1.347(2) | 1.332–1.340 | 1.340–1.342 | |
| 3 | 1.280(2), 1.286(2) | 1.287–1.295 | 1.284–1.286 | |
| 4 | 1.341(2), 1.336(2) | 1.337–1.346 | 1.334–1.347 | |
| 5 | 1.332(3), 1.333(3) | 1.333–1.344 | 1.332–1.338 | |
| 6 | 1.342(3), 1.334(2) | 1.322–1.335 | 1.322–1.335 | |
aLiterature data for the corresponding bond are given as a range including bond lengths in different salts. bBond sequence 1–6 corresponds to that in Table 2.
In salt 9, the bromide ions are held in the crystal structure by hydrogen bonds N15–H15···Br1b [D···A = 3.2774(17) Å, D–H···A = 142°; symmetry code: (b) –x + y + 1/3, −x + 2/3, z + 2/3] and N25–H25···Br2 (D···A = 3.2654(17) Å, D–H···A = 144°). There are also intramolecular hydrogen bonds of the methylene H atoms C2–H2A···N14 [D···A = 3.121(3) Å, D–H···A = 116°].
Theoretical study of structures, UV–vis spectra and the experimental UV–vis spectra of compound 8a
The quantum-chemical study of structure and UV–vis spectra, as well as the experimental study of UV–vis spectra were carried out for compound 8a being the simplest mesoionic compound among the investigated ones in the present work. The atom numbering used in this section corresponds to that shown in Figure 2. The molecule of 8a can be represented by several Lewis structures (Scheme 3).
![[1860-5397-17-34-i3]](/bjoc/content/inline/1860-5397-17-34-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible Lewis structures for the molecule of 8a, with non-Lewis occupancies as % of the total electron density (given in parentheses).
Scheme 3: Possible Lewis structures for the molecule of 8a, with non-Lewis occupancies as % of the total elec...
To find the best Lewis structure for the molecule of 8a, a natural bond orbital (NBO) analysis was applied. In terms of the NBO theory, the "best Lewis structure" is the structure with the lowest non-Lewis occupancy. The NBO search for the molecule of 8a led to the best Lewis structure A (Scheme 3). The high non-Lewis occupancy shows a strong electron delocalization in the molecule of 8a. To study the nature of the electron delocalization for the structure A, an analysis of the interactions between donor Lewis-type NBOs and acceptor non-Lewis NBOs was performed. It showed that the p-type lone pair on the N4 atom is strongly delocalized into vicinal N2–N3* and C5–N5* antibonds, leading to the Lewis structures B and C, correspondingly (Scheme 3). A similar analysis of donor–acceptor interactions for the obtained structures leads to the Lewis structures D and E. Note that the structures B and D with a C5–N5 double bond have only a slightly higher non-Lewis occupancy in comparison with structure A, and hence they are also good Lewis structures. Thus, the structures A, B, and D make a greater contribution to the overall structure of 8a in comparison with structures C and E having a C5–N5 single bond.
As mentioned earlier, compounds 8a–c show solvatochromism. The experimental UV–vis spectra of compound 8a in different solvents are presented in Figure 3a. As can be seen, the experimental spectra in n-hexane and THF, being similar, differ significantly from those in chloroform, methanol, and water. This difference may be due to the following reasons: in case of chloroform, the solvent can form hydrogen bonds with the nitrogen atoms of the 8a molecule, whereas for methanol and water solutions, the solvent can act as a proton donor, and as a result, compound 8a may exist in the 1,3-di-tert-butyl-5-aminotetrazolium cationic form. To confirm this assumption, we calculated the UV–vis spectra of 8a in different solvents for the model structures shown in Figure 4. The model structures in Figure 4b and Figure 4c were built based on the results of our calculations of NPA charges and the molecular electrostatic potential (MESP, Figure 5), showing that the largest negative charge and the deepest minimum of MESP of 8a are located near the exocyclic atom N5, and hence it is the most preferable protonation site in 8a.
![[1860-5397-17-34-3]](/bjoc/content/figures/1860-5397-17-34-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Experimental (a) and TD-tHCTHhyb/6-311+G(2d,p) calculated (b) UV–vis spectra of compound 8a in different solvents. The model structures of 8a, shown in Figure 4 were used in the calculations.
Figure 3: Experimental (a) and TD-tHCTHhyb/6-311+G(2d,p) calculated (b) UV–vis spectra of compound 8a in diff...
![[1860-5397-17-34-4]](/bjoc/content/figures/1860-5397-17-34-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Model structures of 8a used for the calculations of the UV–vis spectra: a) In n-hexane and THF, b) in chloroform, and c) in methanol and water.
Figure 4: Model structures of 8a used for the calculations of the UV–vis spectra: a) In n-hexane and THF, b) ...
![[1860-5397-17-34-5]](/bjoc/content/figures/1860-5397-17-34-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: NPA charges (left) and MESP contour map (right) for the molecule of 8a.
Figure 5: NPA charges (left) and MESP contour map (right) for the molecule of 8a.
The TD-tHCTHhyb/6-311+G(2d,p) calculated UV–vis spectra of compound 8a in n-hexane, THF, chloroform, methanol, and water are presented in Figure 3b. As can be seen, for n-hexane and THF solutions, the experimental and calculated spectra are in good agreement in the case of the model structure in Figure 4a. However, in the case of a chloroform solution, the calculated spectrum agrees with the experimental one only if the formation of a hydrogen bond between the exocyclic N5 atom and the solvent is taken into account (model structure in Figure 4b). For the methanol and water solutions, the agreement between the calculated and experimental spectra is observed only when the protonation of the N5 atom is taken into account (model structure in Figure 4c). For more details, see Supporting Information File 1.
Charge transfer (CT) during excitation plays a key role in many technological applications because CT states correspond to a light-activated electron–hole separation where the positive and negative charges are distant enough to allow their independent collection. In this case, the charge goes from a donor–acceptor (S0) to a donor(+)–acceptor(−) (S1). We have studied, how the nature of the solvent can influence the S0→S1 CT. For this purpose, we calculated plots of the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), the electron density difference between the S1 and S0 states, as well as the S0→S1 CT for compound 8a in the non-polar solvent n-hexane (Figure 6) and in a polar water solution (Figure 7).
![[1860-5397-17-34-6]](/bjoc/content/figures/1860-5397-17-34-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The calculated plots in n-hexane of a) HOMO, b) LUMO, c) electron density difference between the S1 and S0 states, and d) the S0→S1 CT. Green (blue) regions (for c and d) indicate an increase (decrease) in the electron density upon the electronic transition. The red arrow shows the charge transfer.
Figure 6: The calculated plots in n-hexane of a) HOMO, b) LUMO, c) electron density difference between the S1...
![[1860-5397-17-34-7]](/bjoc/content/figures/1860-5397-17-34-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The calculated plots in water of a) HOMO, b) LUMO, c) electron density difference between the S1 and S0 states, and d) the S0→S1 CT. Green (blue) regions (for c and d) indicate an increase (decrease) in the electron density upon electronic transition. The red arrow shows the charge transfer.
Figure 7: The calculated plots in water of a) HOMO, b) LUMO, c) electron density difference between the S1 an...
As it can be seen from the HOMO, LUMO, and electron density difference plots, the S0→S1 electron excitation corresponds to the π→π* transition in n-hexane solution (Figure 6a–c) and to the n→π* transition in water solution (Figure 7a–c). In both cases, the transition is accompanied by a CT from the =N5–H group (donor) to the tetrazole ring (acceptor) (Figure 6d and Figure 7d). For the n-hexane and water solutions, the calculated CT distances are 1.5 and 2.0 Å, respectively. The change in the dipole moment, caused by the excitation, is greater in water being 6.1 D vs 4.9 D in n-hexane. Moreover, the calculated τ index is much greater in the case of the water solution (0.520 Å in water vs 0.017 Å in n-hexane). Note, that a greater positive τ index indicates a stronger charge separation as a result of the electron transition [36]. Hence, in the case of n-hexane, the distribution of the positive and negative charge is almost not separated, and the CT is small. However, for the water solution, the distribution of the positive and negative charge is significantly separated due to a strong CT.
Considering that in polar media the S0→S1 electron excitation corresponds to the n→π* transition, leading to a significant CT from the =N5–H group to the tetrazole ring, we can explain the strong blue shift, observed in methanol and water solution. So, in the ground state, the N5 atom of 8a has a large negative charge (Figure 5) and strongly interacts with polar solvents. Therefore, polar solvents significantly decrease the ground state energy. When the excited state emerges, the strong CT from the =N5–H group to the tetrazole ring (Figure 6c and d) leads to a decrease in the electronic density on the N5 atom. The solvent molecules do not have time to rearrange in order to stabilize the excited state. This results in a lower ground state energy, but not the excited state. Therefore, the energy of S0→S1 transition increases, and a blue shift is observed in polar media.
Conclusion
Mesoionic tetrazolium-5-aminides can be easily prepared by the alkylation of readily available 5-aminotetrazole and its N-alkyl derivatives in a t-BuOH/HClO4 system followed by the treatment of the tetrazolium salts by alkali. The mesoionic compounds show a higher reactivity of the exocyclic N-atom in comparison with the 5-aminotetrazole ones that can be explained by a unique mesoionic system of the tetrazolium-5-aminides which leads up to a 5-amino-group activation. The compounds react with 1,2-dibromoethane and 5-(methylsulfonyl)-1-phenyl-1H-tetrazole and substitute bromine and methylsulfonyl groups giving tetrazolium salts or conjugate aminides. The obtained mesoionic tetrazoles have been characterized by elemental analysis, FTIR, NMR and UV–vis spectroscopy, TGA/DSC analysis, and for 1,3-di-tert-butyltetrazolium-5-aminide, its N,N’-ethylene-bridged bis-derivative and (1,3-di-tert-butyl-1H-tetrazol-3-ium-5-yl)(1-phenyl-1H-tetrazol-5-yl)amide by single crystal X-ray analysis. The structural and spectral features of the tetrazolium-5-aminides were discussed by using quantum-chemical calculations.
Experimental
CAUTION: The prepared 5-iminotetrazoles and their derivatives are energetic compounds with increased sensitivities against heat. Although we had no problems during synthesis, the use of safety equipment such as leather gloves, face shield, and the use of Teflon spatulas is mandatory.
General information
Unless otherwise noted, all reagents were obtained from commercial sources and used without further purification. The UV–vis spectra were recorded on a Merertech SP-8001-6C UV–visible spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker AVANCE 500 MHz spectrometer. IR spectra were recorded on a Bruker Vertex 70 spectrometer in diamond cell accessory. For Raman spectra registration, an Ocean Optics ID RAMAN READER (785 nm) spectrometer was used.
Experimental procedures
The experimental procedures are given in Supporting Information File 1.
Computation details
Calculations of the UV–vis spectra and charge transfer were carried out within density functional theory for ground states and time-dependent density functional theory (TD-DFT) for excited states using the τ-dependent hybrid tHCTHhyb functional [37] with the 6-311+G(2d,p) basis set [38]. The chosen functional allows to predict valence electronic transition energies with high accuracy [39]. However, the tHCTHhyb functional is much less accurate in the calculation of Rydberg electronic transition energies [39]. The geometry of 8a was fully optimized for the ground state in each solvent – n-hexane, THF, chloroform, methanol, water. Solvation effects were considered using the SMD [40] model in terms of Linear Response scheme [41]. The analysis of charge transfer during S0→S1 transition was carried out by using the Multiwfn software [36]. The NBO analysis and MESP calculations were performed using the B3LYP/6-31G(d) level of theory [42].
X-ray structure determination
Single crystal X-ray diffraction data of the mesoionic compounds 8a, 10, 11a, and salt 9 were collected on a SMART Apex II diffractometer using graphite monochromatic Mo Kα radiation (λ = 0.71073 Å) at a temperature of 100 K. The structures were solved by direct methods (SIR2014) [43] and refined on F2 by the full-matrix least squares technique (SHELXL 2014) [44]. The intensities were corrected for absorption. For all compounds, non-hydrogen atoms were refined anisotropically. For compounds 8a, 10, and 11a, all hydrogen atoms were found from the difference Fourier map. For compound 8a, they were refined isotropically; for compounds 10 and 11a, the positions of hydrogen atoms were refined with Uiso(H) = 1.5Ueq(C) for the methyl groups and Uiso(H) = 1.2Ueq(C) for others. For salt 9, the methyl and methylene group H atoms were placed in calculated positions and refined in a “riding model”, with Uiso(H) = 1.5Ueq(C) for the methyl and Uiso(H) = 1.2Ueq(C) for the methylene groups; the hydrogen atoms of N–H groups were found from the difference Fourier map and refined isotropically with Uiso(H) = 1.2Ueq(N). In salt 9, the structure contains large voids, however, the residual electron density in the voids was difficult to model and therefore, the SQUEEZE routine in PLATON [45] was used to remove the contribution of the electron density in the solvent region from the intensity data and the solvent-free model was employed for the final refinement. The solvent contribution was not included in the reported molecular weight and density. Molecular graphics were performed with the programs ORTEP-3 for Windows [46] and PLATON [47]. CCDC deposition numbers for the compounds are 2035296 (8a), 2035297 (10), 2035298 (11a), and 2035299 (9).
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of spectra and calculation results. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Lesnikovich, A. I.; Ivashkevich, O. A.; Levchik, S. V.; Balabanovich, A. I.; Gaponik, P. N.; Kulak, A. A. Thermochim. Acta 2002, 388, 233–251. doi:10.1016/s0040-6031(02)00027-8
Return to citation in text: [1] -
Han, Z. Y.; Zhang, Y. P.; Du, Z. M.; Li, Z. Y.; Yao, Q.; Yang, Y. Z. J. Energ. Mater. 2018, 36, 61–68. doi:10.1080/07370652.2017.1311969
Return to citation in text: [1] -
Dolzhenko, A. V. Heterocycles 2017, 94, 1819–1846. doi:10.3987/rev-17-867
Return to citation in text: [1] -
Ernst, V.; Klapötke, T. M.; Stierstorfer, J. Z. Anorg. Allg. Chem. 2007, 633, 879–887. doi:10.1002/zaac.200700013
Return to citation in text: [1] -
Karaghiosoff, K.; Klapötke, T. M.; Mayer, P.; Sabaté, C. M.; Penger, A.; Welch, J. M. Inorg. Chem. 2008, 47, 1007–1019. doi:10.1021/ic701832z
Return to citation in text: [1] -
Kumbhakarna, N.; Thynell, S. T. Thermochim. Acta 2014, 582, 25–34. doi:10.1016/j.tca.2014.02.014
Return to citation in text: [1] -
Brill, T. B.; Ramanathan, H. Combust. Flame 2000, 122, 165–171. doi:10.1016/s0010-2180(00)00111-5
Return to citation in text: [1] -
Singh, R. P.; Verma, R. D.; Meshri, D. T.; Shreeve, J. M. Angew. Chem., Int. Ed. 2006, 45, 3584–3601. doi:10.1002/anie.200504236
Return to citation in text: [1] -
Tao, G.-H.; Tang, M.; He, L.; Ji, S.-P.; Nie, F.-D.; Huang, M. Eur. J. Inorg. Chem. 2012, 3070–3078. doi:10.1002/ejic.201200065
Return to citation in text: [1] -
Tao, G.-H.; Guo, Y.; Joo, Y.-H.; Twamley, B.; Shreeve, J. M. J. Mater. Chem. 2008, 18, 5524–5530. doi:10.1039/b811506k
Return to citation in text: [1] -
Ollis, W. D.; Ramsden, C. A. Adv. Heterocycl. Chem. 1976, 19, 1–122. doi:10.1016/s0065-2725(08)60230-5
Return to citation in text: [1] -
Moderhack, D. Heterocycles 2016, 92, 185–233. doi:10.3987/rev-15-833
Return to citation in text: [1] -
Bryden, J. H.; Henry, R. A.; Finnegan, W. G.; Boschan, R. H.; McEwan, W. S.; Van Dolah, R. W. J. Am. Chem. Soc. 1953, 75, 4863–4864. doi:10.1021/ja01115a533
Return to citation in text: [1] -
Henry, R. A.; Finnegan, W. G.; Lieber, E. J. Am. Chem. Soc. 1954, 76, 2894–2898. doi:10.1021/ja01640a011
Return to citation in text: [1] -
Araki, S.; Kuzuya, M.; Hamada, K.; Nogura, M.; Ohata, N. Org. Biomol. Chem. 2003, 1, 978–983. doi:10.1039/b211000h
Return to citation in text: [1] -
Araki, S.; Hattori, H.; Ogawa, K.; Kuzuya, M.; Inoue, T.; Yamamura, H.; Kawai, M. J. Chem. Soc., Perkin Trans. 1 2001, 2476–2482. doi:10.1039/b009526p
Return to citation in text: [1] -
Araki, S.; Hattori, H.; Yamamura, H.; Kawai, M. J. Heterocycl. Chem. 2000, 37, 1129–1134. doi:10.1002/jhet.5570370517
Return to citation in text: [1] [2] -
Araki, S.; Yamamoto, K.; Inoue, T.; Fujimoto, K.; Yamamura, H.; Kawai, M.; Butsugan, Y.; Zhou, J.; Eichhorn, E.; Rieker, A.; Huber, M. J. Chem. Soc., Perkin Trans. 2 1999, 985–996. doi:10.1039/a809386e
Return to citation in text: [1] -
Araki, S.; Hattori, H.; Shimizu, N.; Ogawa, K.; Yamamura, H.; Kawai, M. J. Heterocycl. Chem. 1999, 36, 863–867. doi:10.1002/jhet.5570360406
Return to citation in text: [1] -
Araki, S.; Yamamoto, K.; Yagi, M.; Inoue, T.; Fukagawa, H.; Hattori, H.; Yamamura, H.; Kawai, M.; Butsugan, Y. Eur. J. Org. Chem. 1998, 121–127. doi:10.1002/(sici)1099-0690(199801)1998:1<121::aid-ejoc121>3.0.co;2-5
Return to citation in text: [1] -
Bocian, W.; Jaźwiński, J.; Koźmiński, W.; Stefaniak, L.; Webb, G. A. J. Chem. Soc., Perkin Trans. 2 1994, 1327–1332. doi:10.1039/p29940001327
Return to citation in text: [1] -
Jaźwiński, J.; Rozwadowski, Z.; Magiera, D.; Duddeck, H. Magn. Reson. Chem. 2003, 41, 315–323. doi:10.1002/mrc.1182
Return to citation in text: [1] -
Jaźwiński, J.; Staszewska, O.; Stefaniak, L.; Araki, S.; Webb, G. A. J. Mol. Struct. 2000, 523, 103–107. doi:10.1016/s0022-2860(99)00395-6
Return to citation in text: [1] -
Jaźwiński, J. Pol. J. Chem. 1999, 73, 1719–1724.
Return to citation in text: [1] -
Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095
Return to citation in text: [1] [2] [3] [4] [5] -
Voitekhovich, S. V.; Lyakhov, A. S.; Ivashkevich, L. S.; Schleife, F.; Schnorr, R.; Kersting, B.; Gaponik, P. N. Inorg. Chim. Acta 2014, 419, 124–129. doi:10.1016/j.ica.2014.05.008
Return to citation in text: [1] [2] [3] -
Budevich, V. A.; Voitekhovich, S. V.; Ivashkevich, O. A. Synthesis of new mesionic tetrazolium-5-aminide. In Sviridov Readings 2018: International Conference on Chemistry and Chemical Education, Minsk, Belarus, April 10–13, 2018; Vorobyova, T. N.; Vasilevskaya, E. I., Eds.; Krasiko-Print: Minsk, Belarus, 2018; p 87.
Return to citation in text: [1] -
Vaddypally, S.; Tomlinson, W.; O’Sullivan, O. T.; Ding, R.; Van Vliet, M. M.; Wayland, B. B.; Hooper, J. P.; Zdilla, M. J. J. Am. Chem. Soc. 2019, 141, 5699–5709. doi:10.1021/jacs.8b10250
Return to citation in text: [1] [2] [3] [4] -
O'Sullivan, O. T.; Zdilla, M. J. Chem. – Eur. J. 2017, 23, 14138–14142. doi:10.1002/chem.201702696
Return to citation in text: [1] [2] [3] [4] -
Voitekhovich, S. V.; Gaponik, P. N.; Ivashkevich, O. A. Russ. Chem. Rev. 2002, 71, 721–739. doi:10.1070/rc2002v071n09abeh000739
Return to citation in text: [1] -
Ostrovskii, V. A.; Koldobskii, G. I.; Trifonov, R. E. Tetrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K.; Zhdankin, V. V., Eds.; Elsevier: Oxford, Tokyo, 2008; Vol. 6, pp 257–423. doi:10.1016/b978-008044992-0.00517-4
Return to citation in text: [1] -
Klapötke, T. M.; Sabaté, C. M.; Rusan, M. Z. Anorg. Allg. Chem. 2008, 634, 688–695. doi:10.1002/zaac.200700501
Return to citation in text: [1] [2] -
Klapötke, T. M.; Sabaté, C. M. New J. Chem. 2009, 33, 1605–1617. doi:10.1039/b820381d
Return to citation in text: [1] [2] -
Klapötke, T. M.; Sabaté, C. M. Eur. J. Inorg. Chem. 2008, 5350–5366. doi:10.1002/ejic.200800771
Return to citation in text: [1] [2] -
Klapötke, T. M.; Sabaté, C. M.; Penger, A.; Rusan, M.; Welch, J. M. Eur. J. Inorg. Chem. 2009, 880–896. doi:10.1002/ejic.200800995
Return to citation in text: [1] [2] -
Lu, T.; Chen, F. J. Comput. Chem. 2012, 33, 580–592. doi:10.1002/jcc.22885
Return to citation in text: [1] [2] -
Boese, A. D.; Handy, N. C. J. Chem. Phys. 2002, 116, 9559–9569. doi:10.1063/1.1476309
Return to citation in text: [1] -
McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980
Return to citation in text: [1] -
Isegawa, M.; Peverati, R.; Truhlar, D. G. J. Chem. Phys. 2012, 137, 244104. doi:10.1063/1.4769078
Return to citation in text: [1] [2] -
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n
Return to citation in text: [1] -
Cammi, R.; Mennucci, B. J. Chem. Phys. 1999, 110, 9877–9886. doi:10.1063/1.478861
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Burla, M. C.; Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. J. Appl. Crystallogr. 2015, 48, 306–309. doi:10.1107/s1600576715001132
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Spek, A. L. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 9–18. doi:10.1107/s2053229614024929
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/s0021889897003117
Return to citation in text: [1] -
Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x
Return to citation in text: [1]
| 38. | McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980 |
| 39. | Isegawa, M.; Peverati, R.; Truhlar, D. G. J. Chem. Phys. 2012, 137, 244104. doi:10.1063/1.4769078 |
| 39. | Isegawa, M.; Peverati, R.; Truhlar, D. G. J. Chem. Phys. 2012, 137, 244104. doi:10.1063/1.4769078 |
| 1. | Lesnikovich, A. I.; Ivashkevich, O. A.; Levchik, S. V.; Balabanovich, A. I.; Gaponik, P. N.; Kulak, A. A. Thermochim. Acta 2002, 388, 233–251. doi:10.1016/s0040-6031(02)00027-8 |
| 2. | Han, Z. Y.; Zhang, Y. P.; Du, Z. M.; Li, Z. Y.; Yao, Q.; Yang, Y. Z. J. Energ. Mater. 2018, 36, 61–68. doi:10.1080/07370652.2017.1311969 |
| 8. | Singh, R. P.; Verma, R. D.; Meshri, D. T.; Shreeve, J. M. Angew. Chem., Int. Ed. 2006, 45, 3584–3601. doi:10.1002/anie.200504236 |
| 9. | Tao, G.-H.; Tang, M.; He, L.; Ji, S.-P.; Nie, F.-D.; Huang, M. Eur. J. Inorg. Chem. 2012, 3070–3078. doi:10.1002/ejic.201200065 |
| 10. | Tao, G.-H.; Guo, Y.; Joo, Y.-H.; Twamley, B.; Shreeve, J. M. J. Mater. Chem. 2008, 18, 5524–5530. doi:10.1039/b811506k |
| 17. | Araki, S.; Hattori, H.; Yamamura, H.; Kawai, M. J. Heterocycl. Chem. 2000, 37, 1129–1134. doi:10.1002/jhet.5570370517 |
| 28. | Vaddypally, S.; Tomlinson, W.; O’Sullivan, O. T.; Ding, R.; Van Vliet, M. M.; Wayland, B. B.; Hooper, J. P.; Zdilla, M. J. J. Am. Chem. Soc. 2019, 141, 5699–5709. doi:10.1021/jacs.8b10250 |
| 45. | Spek, A. L. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 9–18. doi:10.1107/s2053229614024929 |
| 5. | Karaghiosoff, K.; Klapötke, T. M.; Mayer, P.; Sabaté, C. M.; Penger, A.; Welch, J. M. Inorg. Chem. 2008, 47, 1007–1019. doi:10.1021/ic701832z |
| 6. | Kumbhakarna, N.; Thynell, S. T. Thermochim. Acta 2014, 582, 25–34. doi:10.1016/j.tca.2014.02.014 |
| 7. | Brill, T. B.; Ramanathan, H. Combust. Flame 2000, 122, 165–171. doi:10.1016/s0010-2180(00)00111-5 |
| 25. | Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095 |
| 46. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/s0021889897003117 |
| 4. | Ernst, V.; Klapötke, T. M.; Stierstorfer, J. Z. Anorg. Allg. Chem. 2007, 633, 879–887. doi:10.1002/zaac.200700013 |
| 28. | Vaddypally, S.; Tomlinson, W.; O’Sullivan, O. T.; Ding, R.; Van Vliet, M. M.; Wayland, B. B.; Hooper, J. P.; Zdilla, M. J. J. Am. Chem. Soc. 2019, 141, 5699–5709. doi:10.1021/jacs.8b10250 |
| 43. | Burla, M. C.; Caliandro, R.; Carrozzini, B.; Cascarano, G. L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. J. Appl. Crystallogr. 2015, 48, 306–309. doi:10.1107/s1600576715001132 |
| 29. | O'Sullivan, O. T.; Zdilla, M. J. Chem. – Eur. J. 2017, 23, 14138–14142. doi:10.1002/chem.201702696 |
| 44. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 22. | Jaźwiński, J.; Rozwadowski, Z.; Magiera, D.; Duddeck, H. Magn. Reson. Chem. 2003, 41, 315–323. doi:10.1002/mrc.1182 |
| 23. | Jaźwiński, J.; Staszewska, O.; Stefaniak, L.; Araki, S.; Webb, G. A. J. Mol. Struct. 2000, 523, 103–107. doi:10.1016/s0022-2860(99)00395-6 |
| 24. | Jaźwiński, J. Pol. J. Chem. 1999, 73, 1719–1724. |
| 26. | Voitekhovich, S. V.; Lyakhov, A. S.; Ivashkevich, L. S.; Schleife, F.; Schnorr, R.; Kersting, B.; Gaponik, P. N. Inorg. Chim. Acta 2014, 419, 124–129. doi:10.1016/j.ica.2014.05.008 |
| 15. | Araki, S.; Kuzuya, M.; Hamada, K.; Nogura, M.; Ohata, N. Org. Biomol. Chem. 2003, 1, 978–983. doi:10.1039/b211000h |
| 16. | Araki, S.; Hattori, H.; Ogawa, K.; Kuzuya, M.; Inoue, T.; Yamamura, H.; Kawai, M. J. Chem. Soc., Perkin Trans. 1 2001, 2476–2482. doi:10.1039/b009526p |
| 17. | Araki, S.; Hattori, H.; Yamamura, H.; Kawai, M. J. Heterocycl. Chem. 2000, 37, 1129–1134. doi:10.1002/jhet.5570370517 |
| 18. | Araki, S.; Yamamoto, K.; Inoue, T.; Fujimoto, K.; Yamamura, H.; Kawai, M.; Butsugan, Y.; Zhou, J.; Eichhorn, E.; Rieker, A.; Huber, M. J. Chem. Soc., Perkin Trans. 2 1999, 985–996. doi:10.1039/a809386e |
| 19. | Araki, S.; Hattori, H.; Shimizu, N.; Ogawa, K.; Yamamura, H.; Kawai, M. J. Heterocycl. Chem. 1999, 36, 863–867. doi:10.1002/jhet.5570360406 |
| 20. | Araki, S.; Yamamoto, K.; Yagi, M.; Inoue, T.; Fukagawa, H.; Hattori, H.; Yamamura, H.; Kawai, M.; Butsugan, Y. Eur. J. Org. Chem. 1998, 121–127. doi:10.1002/(sici)1099-0690(199801)1998:1<121::aid-ejoc121>3.0.co;2-5 |
| 21. | Bocian, W.; Jaźwiński, J.; Koźmiński, W.; Stefaniak, L.; Webb, G. A. J. Chem. Soc., Perkin Trans. 2 1994, 1327–1332. doi:10.1039/p29940001327 |
| 27. | Budevich, V. A.; Voitekhovich, S. V.; Ivashkevich, O. A. Synthesis of new mesionic tetrazolium-5-aminide. In Sviridov Readings 2018: International Conference on Chemistry and Chemical Education, Minsk, Belarus, April 10–13, 2018; Vorobyova, T. N.; Vasilevskaya, E. I., Eds.; Krasiko-Print: Minsk, Belarus, 2018; p 87. |
| 13. | Bryden, J. H.; Henry, R. A.; Finnegan, W. G.; Boschan, R. H.; McEwan, W. S.; Van Dolah, R. W. J. Am. Chem. Soc. 1953, 75, 4863–4864. doi:10.1021/ja01115a533 |
| 14. | Henry, R. A.; Finnegan, W. G.; Lieber, E. J. Am. Chem. Soc. 1954, 76, 2894–2898. doi:10.1021/ja01640a011 |
| 40. | Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n |
| 11. | Ollis, W. D.; Ramsden, C. A. Adv. Heterocycl. Chem. 1976, 19, 1–122. doi:10.1016/s0065-2725(08)60230-5 |
| 12. | Moderhack, D. Heterocycles 2016, 92, 185–233. doi:10.3987/rev-15-833 |
| 25. | Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095 |
| 41. | Cammi, R.; Mennucci, B. J. Chem. Phys. 1999, 110, 9877–9886. doi:10.1063/1.478861 |
| 31. | Ostrovskii, V. A.; Koldobskii, G. I.; Trifonov, R. E. Tetrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K.; Zhdankin, V. V., Eds.; Elsevier: Oxford, Tokyo, 2008; Vol. 6, pp 257–423. doi:10.1016/b978-008044992-0.00517-4 |
| 25. | Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095 |
| 47. | Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x |
| 30. | Voitekhovich, S. V.; Gaponik, P. N.; Ivashkevich, O. A. Russ. Chem. Rev. 2002, 71, 721–739. doi:10.1070/rc2002v071n09abeh000739 |
| 37. | Boese, A. D.; Handy, N. C. J. Chem. Phys. 2002, 116, 9559–9569. doi:10.1063/1.1476309 |
| 32. | Klapötke, T. M.; Sabaté, C. M.; Rusan, M. Z. Anorg. Allg. Chem. 2008, 634, 688–695. doi:10.1002/zaac.200700501 |
| 33. | Klapötke, T. M.; Sabaté, C. M. New J. Chem. 2009, 33, 1605–1617. doi:10.1039/b820381d |
| 34. | Klapötke, T. M.; Sabaté, C. M. Eur. J. Inorg. Chem. 2008, 5350–5366. doi:10.1002/ejic.200800771 |
| 35. | Klapötke, T. M.; Sabaté, C. M.; Penger, A.; Rusan, M.; Welch, J. M. Eur. J. Inorg. Chem. 2009, 880–896. doi:10.1002/ejic.200800995 |
| 25. | Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095 |
| 26. | Voitekhovich, S. V.; Lyakhov, A. S.; Ivashkevich, L. S.; Schleife, F.; Schnorr, R.; Kersting, B.; Gaponik, P. N. Inorg. Chim. Acta 2014, 419, 124–129. doi:10.1016/j.ica.2014.05.008 |
| 28. | Vaddypally, S.; Tomlinson, W.; O’Sullivan, O. T.; Ding, R.; Van Vliet, M. M.; Wayland, B. B.; Hooper, J. P.; Zdilla, M. J. J. Am. Chem. Soc. 2019, 141, 5699–5709. doi:10.1021/jacs.8b10250 |
| 29. | O'Sullivan, O. T.; Zdilla, M. J. Chem. – Eur. J. 2017, 23, 14138–14142. doi:10.1002/chem.201702696 |
| 25. | Voitekhovich, S. V.; Gaponik, P. N.; Lyakhov, A. S.; Ivashkevich, O. A. Tetrahedron 2008, 64, 8721–8725. doi:10.1016/j.tet.2008.06.095 |
| 26. | Voitekhovich, S. V.; Lyakhov, A. S.; Ivashkevich, L. S.; Schleife, F.; Schnorr, R.; Kersting, B.; Gaponik, P. N. Inorg. Chim. Acta 2014, 419, 124–129. doi:10.1016/j.ica.2014.05.008 |
| 28. | Vaddypally, S.; Tomlinson, W.; O’Sullivan, O. T.; Ding, R.; Van Vliet, M. M.; Wayland, B. B.; Hooper, J. P.; Zdilla, M. J. J. Am. Chem. Soc. 2019, 141, 5699–5709. doi:10.1021/jacs.8b10250 |
| 32. | Klapötke, T. M.; Sabaté, C. M.; Rusan, M. Z. Anorg. Allg. Chem. 2008, 634, 688–695. doi:10.1002/zaac.200700501 |
| 33. | Klapötke, T. M.; Sabaté, C. M. New J. Chem. 2009, 33, 1605–1617. doi:10.1039/b820381d |
| 34. | Klapötke, T. M.; Sabaté, C. M. Eur. J. Inorg. Chem. 2008, 5350–5366. doi:10.1002/ejic.200800771 |
| 35. | Klapötke, T. M.; Sabaté, C. M.; Penger, A.; Rusan, M.; Welch, J. M. Eur. J. Inorg. Chem. 2009, 880–896. doi:10.1002/ejic.200800995 |
| 29. | O'Sullivan, O. T.; Zdilla, M. J. Chem. – Eur. J. 2017, 23, 14138–14142. doi:10.1002/chem.201702696 |
| 29. | O'Sullivan, O. T.; Zdilla, M. J. Chem. – Eur. J. 2017, 23, 14138–14142. doi:10.1002/chem.201702696 |
© 2021 Budevich et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)