Abstract
A series of γ-oxo esters suitably substituted with various styrene subunits was subjected to samarium diiodide-induced 8-endo-trig cyclizations. Efficacy, regioselectivity and stereoselectivity of these reactions via samarium ketyls strongly depend on the substitution pattern of the attacked alkene moiety. The stereoselectivity of the protonation of the intermediate samariumorganyl is also influenced by the structural features of the substrates. This systematic study reveals that steric and electronic factors exhibited by the alkene and ketone subunits are of high importance for the outcome of these cyclization reactions leading to highly substituted benzannulated cyclooctanol derivatives. In exceptional cases, 7-exo-trig cyclizations to cycloheptanol derivatives have been observed. In examples with high steric hindrance the ketyl–aryl coupling can be a competing process.

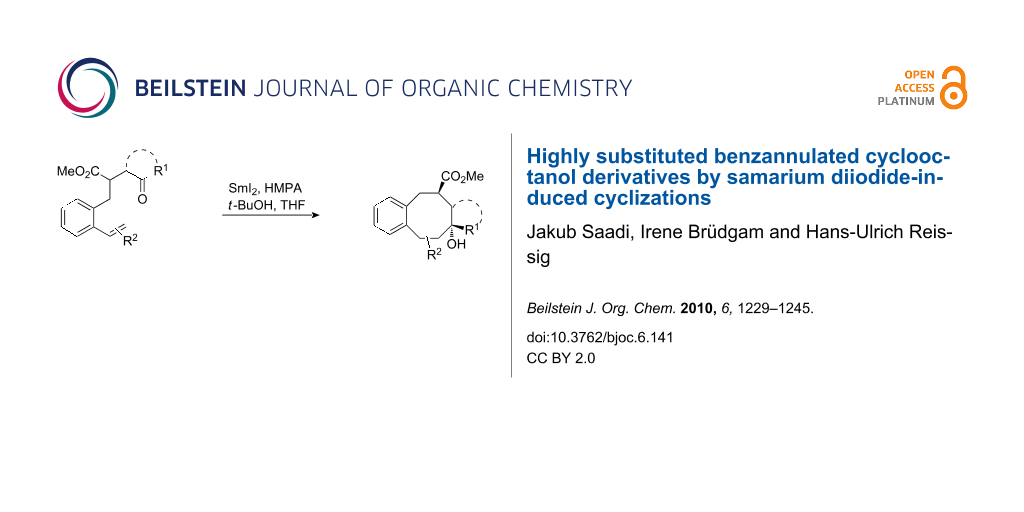
Graphical Abstract
Introduction
Functionalized cyclooctane substructures are frequently found in natural products and pharmacologically significant compounds. Because of their potential biological activity and intriguing geometrical features, the construction of cyclooctanoid frameworks has challenged synthetic organic chemists for a long time [1,2]. This task is quite challenging due to unfavourable enthalpic and entropic factors during the formation of medium-sized rings [3]. Nevertheless, in recent years a range of interesting solutions for the efficient formation of eight-membered rings has been developed [1,2,4-18]. Successful approaches include ring-closing metathesis [4], rearrangements [5], and cycloadditions [6], transition metal-catalyzed cyclizations [7,8], nucleophilic and electrophilic substitution reactions [9] as well as ring expansion reactions [10]. Among these approaches to carbocyclic compounds, samarium diiodide-mediated reactions play an important role and have been described in a number of excellent review articles [19-21] and original publications [22-35]. In our previous reports we have described the efficient synthesis of cyclooctanol and cyclooctenol derivatives by samarium diiodide-induced 8-endo-trig and 8-endo-dig cyclizations of γ-styryl- [20,31,33] and γ-phenylalkynyl-substituted [20,32] ketoesters. Recently, we have also reported our preliminary results on cyclizations of analogous starting materials bearing alkyl and aryl substituents at the styryl double bond, which furnished highly substituted cyclooctanol derivatives (Scheme 1) and in some cases compounds with cycloheptanol substructure [34]. The transformation of A to B proceeds via the samarium ketyl C followed by the 8-endo-trig cyclization to D. Subsequent reduction of the radical D by the second equivalent samarium diiodide and protonation furnishes the cyclooctanol derivative B. The details of this mechanism have been discussed earlier [31]. This method was also successfully applied to the synthesis of cyclic compounds larger than cyclooctane derivatives [35]. Here, we would like to present our detailed results showing the scope and limitations of this method to the synthesis of highly substituted cyclooctanol derivatives and the influence of the substitution pattern of the precursors on the regioselectivity and stereochemical outcome. First, the impact on the reaction by alkyl and aryl substituents at the α-styryl carbon of the substrates will be described, then we discuss the influence of analogous β-styryl substituents.
![[1860-5397-6-141-i1]](/bjoc/content/inline/1860-5397-6-141-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: SmI2-induced cyclizations of styryl-substituted γ-ketoesters A to benzannulated cyclooctanol derivatives B via samarium ketyl C and radical D (HMPA ligands at the samarium are omitted for simplicity in all Schemes, but certainly can play an important role for the outcome).
Scheme 1: SmI2-induced cyclizations of styryl-substituted γ-ketoesters A to benzannulated cyclooctanol deriva...
Results and Discussion
Starting materials were prepared from readily available siloxycyclopropanes in analogy to our previously described modular approach [33]. As a typical example, the synthesis of precursor 4 is depicted in Scheme 2. Cyclopropane 1 [36,37] was deprotonated with LDA and subsequently alkylated with 2-iodobenzyl iodide, furnishing 2 in moderate yield. Intermediate 2 was then treated with triethylamine trihydrofluoride, furnishing 2-iodobenzyl-substituted γ-ketoester 3. The following palladium-catalyzed cross-coupling [38,39] with potassium 2-propenyl trifluoroborate afforded the cyclization precursor 4 in very good yield.
![[1860-5397-6-141-i2]](/bjoc/content/inline/1860-5397-6-141-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three-step synthesis of precursor 4 starting from siloxycyclopropane derivative 1.
Scheme 2: Three-step synthesis of precursor 4 starting from siloxycyclopropane derivative 1.
The cyclization reactions were generally performed with 2.2 equiv of samarium diiodide in THF and in the presence of 18 equiv of HMPA and 2 equiv of tert-butanol. HMPA is crucial for the success of most of the described cyclizations because of its unique influence on the reactivity of samarium diiodide [40-44]. For control experiments and an add-on to our previous observations [33], we started our experiments with the cyclization of ketoesters 5a/b containing a cycloheptanone subunit. Here only the unlike-configured [45] 5a furnished the expected benzannulated tricyclic compound 6 in moderate yield (Scheme 3). The like-configured precursor 5b is unable to arrange the reacting moieties (carbonyl group and alkene) in appropriate proximity while retaining an energetically favourable conformation of the molecule. The impact of the relative configuration of cyclic ketones as starting materials on the 8-endo-trig process is in full agreement with our previous observations for the corresponding cyclohexanone derivatives [33].
![[1860-5397-6-141-i3]](/bjoc/content/inline/1860-5397-6-141-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Attempted cyclizations of diastereomeric cycloheptanone derivatives 5a and 5b.
Scheme 3: Attempted cyclizations of diastereomeric cycloheptanone derivatives 5a and 5b.
Because of the observed sensitivity of this reaction to steric factors, it was necessary to study the tolerance and limitations for the substitution pattern of the starting materials in more detail. At first, the influence of a methyl substituent at the α-styryl carbon was investigated. Cyclization of the unlike-configured cyclohexanone derivative 7a afforded the expected tricyclic cyclooctanol derivative 8 in good yield and excellent trans-stereoselectivity (Scheme 4). The terms cis and trans refer in this report to the position of the methoxycarbonyl group and the hydroxyl group. In general, cis-configured products directly undergo a subsequent cyclization to the corresponding γ-lactones. It is noteworthy that the additionally introduced stereogenic centre bearing the methyl group is also generated stereoselectively.
![[1860-5397-6-141-i4]](/bjoc/content/inline/1860-5397-6-141-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Samarium diiodide-induced cyclization of γ-ketoester 7a to tricyclic compound 8.
Scheme 4: Samarium diiodide-induced cyclization of γ-ketoester 7a to tricyclic compound 8.
In part, the configuration of 8 was assigned based on the relative configuration of the starting material. The lack of lactone formation indicates that the bridgehead hydroxyl group is trans located with respect to the methoxycarbonyl group. The configuration at the methyl-substituted stereocenter was assigned based on NOESY-correlations and careful comparison of spectroscopic data with those of related compounds. The analogous cyclopentanone-derived starting material was also tested in the cyclization reaction; however, it afforded a complex mixture of products.
Cyclization of acyclic starting material 4, bearing a methyl substituent at the α-styryl carbon, furnished a mixture of trans- and cis-cyclization products 9 and 10 in very good combined yield, but practically no stereochemical preference. The precursor 11, bearing the more bulky iso-propyl ketone subunit, provided cyclooctanol derivatives 12 and 13 with similarly good combined yield and a clear preference for trans-product 12 (Scheme 5).
![[1860-5397-6-141-i5]](/bjoc/content/inline/1860-5397-6-141-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Samarium diiodide-induced cyclizations of methyl ketone 4 and iso-propyl ketone 11.
Scheme 5: Samarium diiodide-induced cyclizations of methyl ketone 4 and iso-propyl ketone 11.
The relative configurations of lactone-bridged compounds 10 and 13 were deduced from NOESY-correlations and the assumption was confirmed that the five-membered lactone bridge, which is quite constrained and rigid, can be formed only when carboxyl and hydroxyl groups are at the same face of the eight-membered ring (Figure 1). This analysis was further supported by high similarity of NMR-spectroscopic data (1H and 13C NMR shifts and relative signal patterns) with those of compound 17a and at the same time rather big differences to those of the epimeric compound 17b.
![[1860-5397-6-141-1]](/bjoc/content/figures/1860-5397-6-141-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: NOESY-correlation for compound 10.
Figure 1: NOESY-correlation for compound 10.
The NOESY-experiments also indicated the proton correlations across the eight-membered ring in compounds 9 and 12. The assignment was further supported by the correlations between the protons at carbons with one substituent at the ring and their neighbouring protons. The cis-vicinal protons indicated strong correlations, while the trans-vicinal protons, where the dihedral angle was close to 180°, show none or almost no correlations between each other (Figure 2).
![[1860-5397-6-141-2]](/bjoc/content/figures/1860-5397-6-141-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: NOESY-correlation for compound 9.
Figure 2: NOESY-correlation for compound 9.
In cyclizations of the (2-propenyl)phenyl-substituted ketoesters, trans-products are preferred; however the cis/trans stereoselectivity was generally lower than that observed for the cyclizations of their styrene analogues [33]. It seems that the previously observed correlation between the bulkiness of a ketone subunit and the increase in stereoselectivity is also effective here. This result is in accordance to the proposed eight-membered pseudo-chair-like transition structure [33], where the methoxycarbonyl group occupies a preferred pseudo-equatorial position and the ketyl substituent is competing with the bulky samariumoxy group for the other pseudo-equatorial position. Therefore, ketones with bulkier substituents preferentially react through a transition structure A (Scheme 6) leading to the trans-cyclization product. Remarkably, the orientation of the methyl group at the newly formed third stereogenic centre, which arises during the protonation of the samariumorganyl intermediate, was always trans to the hydroxyl group, arising from the ketone moiety. This interesting observation indicates that the protonation is highly stereoselective and the protons are coming from the same face of the ring where the samariumoxy group is situated after the completed cyclization. Since samarium(III) is very oxophilic, it is possible that the samariumoxy group coordinates tert-butanol, which is then a more acidic proton source (intermediates B and C, Scheme 6). This template effect would then be responsible for the stereoselective protonation governed by the samariumoxy moiety. An intermediate similar to C (covalent C–Sm bond or contact ion pair) may be a plausible alternative, which is then protonated under retention by the proton source [46].
![[1860-5397-6-141-i6]](/bjoc/content/inline/1860-5397-6-141-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Assumed transition structures and intermediates A, B, or C for the cyclizations of (2-propenyl)phenyl-substituted ketones 4 and 11 leading to products of type D (HMPA ligands at the samarium are omitted for simplicity).
Scheme 6: Assumed transition structures and intermediates A, B, or C for the cyclizations of (2-propenyl)phen...
The introduction of two additional methyl substituents between the ketone and ester moieties in the bulky diisopropyl ketone derived substrate 14, suppressed the 8-endo-trig cyclization completely and only the fragmentation product 15 was isolated from the reaction mixture (Scheme 7). It is not clear whether the mechanism of SmI2-induced fragmentation of 1,4-dicarbonyl compounds is of anionic or radical nature, but it has been observed in several cases [33,47-50]. It is possible that, due to the high steric hindrance of the initially formed samarium ketyl, the coupling to the alkene is very slow. The ketyl or the anion formed by its further reduction with SmI2 fragments to the samarium enolate of diisopropyl ketone and the radical or anion stabilized by the methoxycarbonyl group. Very surprisingly in this reaction, small amounts of the n-propyl ester of 15 were also found. At this moment, the origin of the n-propyl group is not clear.
![[1860-5397-6-141-i7]](/bjoc/content/inline/1860-5397-6-141-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reductive fragmentation of highly hindered ketoester 14.
Scheme 7: Reductive fragmentation of highly hindered ketoester 14.
The modulation analysis of the electronic properties of the reacting alkene was evaluated by the introduction of a phenyl substituent to the α-styryl position. The samarium diiodide-induced cyclization of ketoester 16 afforded two epimeric lactone bridged cis-products, 17a and 17b, in ca. 1.7:1 ratio and in good combined yield (Scheme 8).
![[1860-5397-6-141-i8]](/bjoc/content/inline/1860-5397-6-141-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Samarium diiodide-induced cyclization of phenyl-substituted substrate 16 leading to lactones 17a and 17b.
Scheme 8: Samarium diiodide-induced cyclization of phenyl-substituted substrate 16 leading to lactones 17a an...
The relative configuration of 17b was unambiguously assigned by X-ray crystal structure (Figure 3) [51]. Assuming that the lactone bridge is formed only when carboxyl and hydroxyl groups are at the same face of the eight-membered ring, the configuration of 17a with the inverted phenyl-substituted stereocenter was assigned as the only alternative to 17b.
![[1860-5397-6-141-3]](/bjoc/content/figures/1860-5397-6-141-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure (Diamond [52]) of compound 17b.
Figure 3: Molecular structure (Diamond [52]) of compound 17b.
The exclusive cis-selectivity of cyclization, observed in this case, may be attributed to a high level of the steric repulsions between the bulky phenyl group at the alkene moiety and the iso-propyl group at the samarium ketyl in the usually preferred trans-transition structure (compare A, Scheme 6). It is unclear, why the protonation of the samariumorganyl species, which is additionally stabilized by the phenyl substituent, occurred with lower stereochemical preference in this case. However, if an intermediate such as C (Scheme 6) is also involved in this transformation a less covalent C-Sm bond may lead to a decreased stereoselectivity of the protonation step.
We have subsequently investigated the influence of alkyl substituents at the β-styryl carbon. In this series, (E)-1-propenyl-substituted acyclic ketones 18, 21, and 24 gave the expected trans-cyclooctanol derivatives 19, 22, and 25 in moderate to good yields and with excellent stereoselectivities (Scheme 9). In case of cyclization of methyl-substituted ketone 18, the side-product 20 was obtained as a result of a ketyl–aryl coupling [20,53-58] in addition to the desired cyclooctanol derivative 19. Also minor amounts of fragmentation product 23 were detected in the cyclization of ethyl ketone 21 (Scheme 9). These two observations indicate that the transition structures of the cyclizations leading to eight-membered rings are more constrained and that the reactions are slower than those where a terminally unsubstituted alkene is attacked.
![[1860-5397-6-141-i9]](/bjoc/content/inline/1860-5397-6-141-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Samarium diiodide-induced cyclizations of (E)-(1-propenyl)phenyl-substituted γ-ketoesters 18, 21, and 24.
Scheme 9: Samarium diiodide-induced cyclizations of (E)-(1-propenyl)phenyl-substituted γ-ketoesters 18, 21, a...
The assignment of the configuration of product 20 is based on that of numerous earlier described examples of SmI2-induced ketyl–aryl coupling products [57]. The configurations of products 19, 22, and 25 were assigned on the basis of NOESY-correlations.
The transition structure for the SmI2-mediated 8-endo-trig cyclizations of these (E)-1-propenyl-substituted substrates is proposed in accordance to the previously postulated eight-membered pseudo-chair-like transition structures (Figure 4). The methoxycarbonyl group and substituent R at the ketyl moiety are both occupying preferred pseudo-equatorial positions. In our drawing, the olefin approaches the samarium ketyl from the backside perpendicularly to the bulky samariumoxy group. The methyl substituent at the alkene moiety prefers a staggered position in between the bulky samariumoxy group and substituent R.
![[1860-5397-6-141-4]](/bjoc/content/figures/1860-5397-6-141-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Proposed transition structure for the cyclization of (E)-1-propenyl-substituted substrates (HMPA ligands and proton donors ROH at the samarium are omitted for simplicity).
Figure 4: Proposed transition structure for the cyclization of (E)-1-propenyl-substituted substrates (HMPA li...
Another indication of the considerable steric repulsion introduced by the methyl substituent at the β-styryl position is provided by the cyclizations of sterically more constrained cyclohexanone derivatives 26a and 26b (Scheme 10). In these two cases both the usually preferred unlike-configured precursor 26a and the disfavoured like-configured 26b gave only low quantities of cyclooctanol products, 27 and 29, respectively, along with the recovered starting material and either the ketyl-aryl coupling product 28 or the fragmentation product 23 (Scheme 10).
![[1860-5397-6-141-i10]](/bjoc/content/inline/1860-5397-6-141-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Attempted samarium diiodide-induced cyclizations with (E)-1-propenyl-substituted precursors 26a and 26b.
Scheme 10: Attempted samarium diiodide-induced cyclizations with (E)-1-propenyl-substituted precursors 26a and ...
The stereochemical assignments for products 27 and 29 are based on the relative configuration of their precursors. The existence or lack of lactone formation allows for assigning the configuration of the bridgehead carbon bearing the hydroxyl group. The assignment of the centre with the methyl group is based on a strong NOESY-correlation between the methyl protons and the bridgehead proton. It is noteworthy that the stereoselectivity of the cyclization for cyclic ketones is clearly controlled by the relative configuration of the precursors and that the hydroxyl group is generated trans to the vicinal substituents. The reactivity of the sterically more demanding β-styryl-substituted substrates was strongly retarded due to rigidity and bulkiness of the integrated cyclohexane ring, which in the previous cyclization examples afforded cyclooctanol products with good yield and excellent stereoselectivity.
The cyclopentanone-derived analogues of 26a/b were also examined in this samarium diiodide-promoted transformation, but among the isolated products only the starting materials could be unequivocally identified. Not surprisingly, the (E)-1-propenyl analogue of the bulky diisopropyl ketone-derived substrate 14 only afforded the corresponding fragmentation product. In this case again small amounts of the n-propyl ester were detected (compare Scheme 7).
The influence of the configuration of the reacting alkene was also studied. The cyclization of (Z)-1-propenyl-substituted 30, which is isomeric to already examined (E)-1-propenyl-ketoester 24, afforded the corresponding cyclooctanol 31 with considerably lower efficacy (Scheme 11, compare Scheme 9). The reaction mixture afforded considerable amounts of fragmentation product 32 along with unchanged starting material.
![[1860-5397-6-141-i11]](/bjoc/content/inline/1860-5397-6-141-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Attempted samarium diiodide-induced cyclization of (Z)-1-propenyl-substituted precursor 30.
Scheme 11: Attempted samarium diiodide-induced cyclization of (Z)-1-propenyl-substituted precursor 30.
The configuration of product 31 is based on the comparison with the isomeric product 25. Analysis of a possible transition structure analogous to that proposed for the cyclization of (E)-1-propenyl-substituted γ-ketoesters (compare Figure 4) reveals that the Z-configured alkene has to arrange the methyl substituent in a highly unfavourable endo-cyclic position of the newly formed ring. This apparently causes high steric repulsion and thus strongly suppressed reactivity.
In order to further define the scope and limitations of the samarium diiodide-induced reaction we then studied the cyclization of β-styryl-substituted γ-ketoester 33. This precursor has the preferred (E)-configured alkene substructure, but is equipped with branched substituents at the ketone and alkene moieties (Scheme 12). The starting material was completely consumed in this reaction, affording a moderate yield of the eight-membered carbocycle 34 together with a fairly high amount of fragmentation product 35. The obtained product ratio suggests that in this example, the cyclization is roughly two times slower than the fragmentation. When a phenyl group was introduced to the β-styrene position the cyclization of γ-ketoester 36 mainly afforded the desired cyclooctanol derivative 37, along with several undefined side products in low quantities and 20% of unconsumed starting material (Scheme 12). It has to be emphasized here, that the 1,2-diarylalkene unit can potentially react with the samarium ketyl at both carbon atoms affording two different stabilized benzylic radicals. Therefore it is an interesting observation that the 8-endo-trig cyclization mode is preferred over the possible 7-exo-trig mode for the reaction of compound 36.
![[1860-5397-6-141-i12]](/bjoc/content/inline/1860-5397-6-141-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Samarium diiodide-induced cyclizations of γ-ketoesters 33 and 36.
Scheme 12: Samarium diiodide-induced cyclizations of γ-ketoesters 33 and 36.
The configurations of products 34 and 37 are assigned with the aid of NOESY-correlations and by comparison with the NMR-spectroscopic data of analogous compounds obtained after cyclizations of (E)-1-propenyl-substituted γ-ketoesters.
In contrary to the reaction of acyclic ketone 36, stilbenyl-substituted cyclohexanone derivatives 38a and 38b both preferred to undergo a 7-exo-trig cyclization, yielding tricyclic cycloheptanol derivative 39 and tetracyclic lactone 40, respectively (Scheme 13). Similarly as in cyclizations of the cyclohexanone-derived ketoesters with terminally substituted alkenes 26a/b, the 8-endo-trig cyclization pathway may be quite hampered in the cases of 38a and 38b. However, due to the radical stabilizing properties of the terminal phenyl substituent, the 7-exo-trig pathway is now possible. It is probable that the restricted conformational flexibility of the cyclic ketones leads to higher steric and torsional strain in an alternative eight-membered transition structure. This assumption is supported by the observation that conformationally more flexible acyclic γ-ketoesters, such as 36, prefer the 8-endo-trig cyclization mode. Furthermore, the like-configured starting material 38b, which is disfavoured in 8-endo-trig cyclizations, afforded the cycloheptanol derivative with even better efficacy than the unlike-configured 38a.
![[1860-5397-6-141-i13]](/bjoc/content/inline/1860-5397-6-141-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Samarium diiodide-induced cyclizations of diastereomeric stilbenyl-substituted γ-ketoesters 38a and 38b.
Scheme 13: Samarium diiodide-induced cyclizations of diastereomeric stilbenyl-substituted γ-ketoesters 38a and ...
The constitution and the relative configuration of compound 40, featuring a tetracyclic lactone-bridged core, were unambiguously determined by an X-ray crystal structure (Figure 5) [59]. The constitution of compound 39 was assigned as a cycloheptanol derivative based on the similarities of its 13C NMR data with those of 40 rather than those of the analogous eight-membered carbocycles. The configuration of 39 was assigned by using the following arguments: the relative configuration of the unlike-configured precursor should be transferred to the product and the bridgehead hydroxyl group should be in trans-relationship to the methoxycarbonyl group since no lactone formation was observed. The configuration of the stereogenic centre with the phenyl group of 39 is based on NOESY-correlations between the exocyclic benzylic protons and the bridgehead proton.
![[1860-5397-6-141-5]](/bjoc/content/figures/1860-5397-6-141-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure (Diamond [52]) of compound 40.
Figure 5: Molecular structure (Diamond [52]) of compound 40.
An attempt to cyclize γ-ketoester 41 featuring a β-dialkyl-substituted styryl moiety furnished only minor amounts of the ketyl–aryl coupling products 42 and 43 (Scheme 14). Similar to some other reactions leading mainly to side products, trace amounts of n-propyl ester of 42 of unknown origin were detected. Although the precursor was completely consumed, the missing material could not be identified. The relative configuration of the hexahydronaphthalene derivatives was assigned by the comparison of spectroscopic data with that of the previously described analogous compounds [57].
![[1860-5397-6-141-i14]](/bjoc/content/inline/1860-5397-6-141-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Attempted cyclization of β-dialkyl-substituted styrene derivative 41.
Scheme 14: Attempted cyclization of β-dialkyl-substituted styrene derivative 41.
Conclusion
Samarium diiodide-mediated 8-endo-trig cyclizations of styryl-substituted γ-ketoesters bearing alkyl or aryl substituents at the α- or β-styryl carbon were systematically studied. The stereoselectivity of these transformations was strongly influenced by the steric bulk at the ketone and alkene moieties. Acyclic γ-ketoesters such as A cyclized very efficiently, affording mixtures of cis- and trans-products B and C with improving cis/trans stereoselectivity by increase of the size of the ketone substituents (Scheme 15).
![[1860-5397-6-141-i15]](/bjoc/content/inline/1860-5397-6-141-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Typical products of samarium diiodide-induced 8-endo-trig cyclizations of α-styryl-substituted γ-ketoesters.
Scheme 15: Typical products of samarium diiodide-induced 8-endo-trig cyclizations of α-styryl-substituted γ-ke...
γ-Ketoesters with substituents at the β-styryl carbon such as D cyclized with similar efficiency, affording in most cases the 8-endo-trig-cyclization products E. Only cyclohexanone derived γ-ketoesters F preferred the 7-exo-trig over the 8-endo-trig-cyclization mode (Scheme 16).
![[1860-5397-6-141-i16]](/bjoc/content/inline/1860-5397-6-141-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Typical products of samarium diiodide-induced 8-endo-trig cyclizations of β-styryl-substituted γ-ketoesters D and of 7-exo-trig cyclizations of β-styryl-substituted γ-ketoesters F.
Scheme 16: Typical products of samarium diiodide-induced 8-endo-trig cyclizations of β-styryl-substituted γ-ke...
A typical side reaction, which was observed for methyl ketones or cyclohexanone derivatives, was the ketyl-aryl coupling reaction leading to hexahydronaphthalene derivatives in low yields.
In conclusion, the intramolecular 8-endo-trig samarium ketyl-alkene coupling reaction is a flexible tool for the construction of highly substituted cyclooctanol derivatives. A careful design of the precursors following the rules determined in this study allows the synthesis of functionalized benzannulated cyclooctanol derivatives with a high degree of regio- and stereocontrol.
Experimental
General: All reactions were carried out under argon in flame-dried flasks, and the components were added by syringe. All solvents were dried by standard methods. Thin layer chromatography (TLC) was carried out on commercial Polygram Sil G/UV254 or Polygram Alox N/UV254 (Macherey & Nagel). Column chromatography was performed with 70–230 mesh silica gel (Merck) or neutral aluminium oxide (activity grade III; Fluka or Merck). Unless stated otherwise, 1H NMR and 13C NMR spectra were determined with Bruker AC 200, AC 300, DRX 500, Avance III 700 or Jeol Eclipse 500 instruments in CDCl3 solution. The chemical shifts refer to TMS or to the CDCl3 signal (δH = 7.26 ppm. δC = 77.16 ppm). IR spectra were measured with a Nicolet 205 5SXC FTIR-interferometer, equipped with a DTGS-detector. Melting points are uncorrected and determined with a melting point microscope Büchi 510. MS and HRMS analyses were performed with Finnigan MAT 711 (EI = 80 eV, 8 kV), MAT 95 (EI = 70 eV), MAT CH7A (EI = 80 eV, 3 kV) and CH5DF (FAB = 80 eV, 3 kV) instruments. Elemental analyses were performed with Perkin-Elmer and Vario EL Elementar analytical equipment.
Preparation of all cyclization precursors analogously to known procedures is described in Supporting Information File 1.
SmI2-induced cyclization
General procedure A [60,61]: Samarium metal (2.4 equiv) and 1,2-diiodoethane (2.2 equiv) were placed under a flow of argon in a flame-dried, two-necked round-bottomed flask containing a magnetic stirring bar and a septum inlet. THF (12 mL/mmol of 1,2-diiodoethane) was added and the mixture was vigorously stirred at rt for 2 h. HMPA (18 equiv) was added to this solution of SmI2 (2.2 equiv), and after 10 min of stirring, a solution of the substrate (1 equiv) and t-BuOH (2 equiv) in THF (40 mL/mmol of substrate) was added over 2 h. The mixture was stirred at rt for 16 h and quenched with satd. aqueous NaHCO3 solution (20 mL/mmol of substrate). The phases were separated, and the aqueous layer was extracted with diethyl ether (3 × 15 mL/mmol of substrate). The combined organic layers were washed with water and brine (10 mL/mmol of substrate) and dried (Na2SO4).
General procedure B [62,63]: Samarium metal (2.48 g, 16.5 mmol) and iodine (3.81 g, 15.0 mmol) were placed under a flow of argon in a flame-dried, 250 mL round-bottomed flask containing a magnetic stirring bar. Then 150 mL of dry and oxygen-free THF was added and the mixture was covered from light, and stirred at rt for 24 h to give a 0.1 M solution of SmI2 in THF. This solution was stored in the dark under argon at rt and aliquots were transferred with a syringe (2.2 equiv) into the two-necked reaction flask, and the solution was then used as described in General procedure A.
SmI2-induced cyclization of 5a
General procedure B: 5a (0.300 g, 1.00 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 6 (0.126 g, 42%) as colourless crystals (mp 126–127 °C) and 5a (0.069 g, 23%).
Methyl (6RS,6aSR,11aRS)-11a-hydroxy-6,6a,7,8,9,10,11,11a,12,13-decahydro-5H-benzo[a]cyclohepta[e][8]annulene-6-carboxylate (6): 1H NMR (CDCl3, 700 MHz): δ = 1.11–1.15, 1.20–1.33, 1.52–1.75, 1.77–1.81 (4 m, 2 H, 3 H, 6 H, 1 H, 6a-H, 7-H, 8-H, 9-H, 10-H, 11-H, OH), 1.88 (ddd, J = 2.2, 8.0, 14.4 Hz, 1 H, 12-H), 2.06 (ddd, J = 2.6, 11.6, 14.4 Hz, 1 H, 12-H), 2.68 (ddd, J = 2.6, 8.0, 14.0 Hz, 1 H, 13-H), 2.85 (dd, J = 4.6, 14.2 Hz, 1 H, 5-H), 3.01 (ddd, J = 4.6, 5.2, 10.6 Hz, 1 H, 6-H), 3.18 (ddd, J = 2.2, 11.6, 14.0 Hz, 1 H, 13-H), 3.62 (dd, J = 5.2, 14.2 Hz, 1 H, 5-H), 3.69 (s, 3 H, CO2Me), 6.89–6.91, 7.12–7.18 (2 m, 1 H, 3 H, Ar) ppm. 13C NMR (CDCl3, 175 MHz): δ = 23.6, 29.1, 29.4, 29.5 (4 t, C-7, C-8, C-9, C-10), 31.5 (t, C-13), 34.9 (t, C-5), 45.0 (d, C-6a), 46.9, 47.0 (2 t, C-11, C-12), 51.4 (d, C-6), 126.1, 127.0, 129.1, 130.5, 136.8, 142.0 (4 d, 2 s, Ar), 51.2, 176.1 (q, s, CO2Me) ppm. Signal for C-11a is not clearly visible. IR (KBr): ![[Graphic 1]](/bjoc/content/inline/1860-5397-6-141-i17.svg?max-width=637&scale=1.063638) = 3500 (O–H), 3060–2855 (=C–H, C–H), 1715 (C=O) cm–1. C19H26O3 (302.4): calcd. C 75.46, H 8.67; found: C 75.36, H 8.78.
= 3500 (O–H), 3060–2855 (=C–H, C–H), 1715 (C=O) cm–1. C19H26O3 (302.4): calcd. C 75.46, H 8.67; found: C 75.36, H 8.78.
SmI2-induced cyclization of 7a
General Procedure A: 7a (0.296 g, 0.99 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) then HPLC (iso-propanol/hexane 7:93) to furnish 8 (0.187 g, 63%) as colourless crystals (mp 74–75 °C).
Methyl (4aSR,5RS,11RS,12aRS)-12a-hydroxy-11-methyl-1,2,3,4,4a,5,6,11,12,12a-decahydro-dibenzo[a,e][8]annulene-5-carboxylate (8): 1H NMR (CDCl3, 500 MHz): δ = 1.14–1.18 (m, 2 H, 3-H, 4-H), 1.28 (d, J = 7.0 Hz, 3 H, 11-Me), 1.30–1.34, 1.38–1.46, 1.54–1.58, 1.63–1.70 (4 m, 1 H, 3 H, 1 H, 2 H, 1-H, 2-H, 3-H, 4-H, 4a-H), 1.96 (dd, J = 4.9, 15.2 Hz, 1 H, 12-H), 2.08 (dd, J = 11.5, 15.2 Hz, 1 H, 12-H), 2.63 (ddd, J = 4.7, 5.8, 10.6 Hz, 1 H, 5-H), 3.13–3.20 (m, 2 H, 6-H), 3.35 (dqd, J = 4.9, 7.0, 11.5 Hz, 1 H, 11-H), 3.73 (s, 3 H, CO2Me), 6.98–7.00, 7.12–7.21 (2 m, 1 H, 3 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz): δ = 21.3 (t, C-2), 24.3 (q, 11-Me), 24.9 (t, C-3), 29.6 (t, C-4), 33.7 (d, C-11), 34.1 (t, C-6), 44.3 (t, C-1), 45.0 (d, C-4a), 53.2 (d, C-5), 54.0 (t, C-12), 73.2 (s, C-12a), 126.4, 127.4, 127.6, 131.0, 137.3, 144.8 (4 d, 2 s, Ar), 51.7, 176.6 (q, s, CO2Me) ppm. IR (KBr): = 3510 (br, O-H), 3060–2860 (=C-H, C-H), 1725 (C=O) cm−1. MS (EI, 70 eV): m/z (%) = 302 (21) [M]+, 284 (100), 224 (64), 169 (57), 131 (53), 117 (60), 91 (44), 43 (46). C19H26O3 (302.4): calcd. C 75.46, H 8.67; found: C 75.23, H 8.71.
SmI2-induced cyclization of 4
General Procedure A: 4 (0.200 g, 0.77 mmol), SmI2 (1.70 mmol), HMPA (2.43 mL, 13.8 mmol) and t-BuOH (0.114 g, 1.54 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 9 (0.080 g, 40%) as a colourless oil and 10 (0.067 g, 38%) as colourless crystals (mp 130–132 °C).
Methyl (6RS,8RS,10RS)-8-hydroxy-8,10-dimethyl-5,6,7,8,9,10-hexahydro-benzo-[8]annulene-6-carboxylate (9): Compound 9 shows temperature-dependent NMR spectra. At rt some signals appear broad; however, for measurements at 55 °C, the signals are more clearly seen. 1H NMR (CDCl3, 500 MHz, 55 °C): δ = 1.20 (s, 3 H, 8-Me), 1.33 (dd, J = 11.5, 14.7 Hz, 1 H, 7-H), 1.33 (d, J = 7.1 Hz, 3 H, 10-Me), 1.62 (dd, J = 11.4, 14.3 Hz, 1 H, 9-H), 1.67 (dd, J = 3.8, 14.7 Hz, 1 H, 7-H), 1.79–1.83 (m, 1 H, 9-H), 3.10 (dddd, J = 2.9, 3.8, 7.5, 11.5 Hz, 1 H, 6-H), 3.16 (dd, J = 2.9, 13.9 Hz, 1 H, 5-H), 3.36–3.43 (m, 1 H, 10-H), 3.43 (dd, J = 7.5, 13.9 Hz, 1 H, 5-H), 3.68 (s, 3 H, CO2Me), 6.97–6.99, 7.06–7.10, 7.17–7.25 (3 m, 1 H, 1 H, 2 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz, 55 °C): δ = 23.2 (q, 10-Me), 30.2 (d, C-10), 32.3 (t, C-5), 36.0 (q, 8-Me), 36.5 (t, C-7), 42.2 (d, C-6), 55.1 (t, C-9), 71.5 (s, C-8), 125.1, 125.9, 127.1, 129.6, 136.5, 146.3 (4 d, 2 s, Ar), 51.7, 176.1 (q, s, CO2Me) ppm. IR (neat): = 3500 (br, O-H), 3100–2840 (=C-H, C-H), 1715 (C=O) cm–1. HRMS (ESI) calcd for C16H22O3: [M+H]+ = 217.1255, [M+Na]+ = 239.1074, [M+K]+ = 255.0813; found: 217.1267, 239.1095, 255.0844.
(2RS,5SR,7SR)-5,7-Dimethyl-1,5,6,7-tetrahydro-2,5-methano-4-benzoxonin-3(2H)-one (10): 1H NMR (CDCl3, 500 MHz): δ = 1.34 (d, J = 6.9 Hz, 3 H, 7-Me), 1.36 (s, 3 H, 5-Me), 1.43 (dd, J = 1.1, 13.9 Hz, 1 H, 12-H), 1.55 (dd, J = 11.3, 14.6 Hz, 1 H, 6-H), 1.74–1.79 (m, 1 H, 12-H), 2.05–2.09 (m, 1 H, 6-H), 2.76 (dqd, J = 1.3, 6.9, 11.3 Hz, 1 H, 7-H), 3.15 (dddd, J = 1.1, 2.5, 10.3, 12.8 Hz, 1 H, 2-H), 3.20 (dd, J = 12.8, 14.5 Hz, 1 H, 1-H), 3.31 (dd, J = 2.5, 14.5 Hz, 1 H, 1-H), 7.00–7.02, 7.15–7.18, 7.28–7.36 (3 m, 1 H, 1 H, 2 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 22.5 (q, 7-Me), 29.6 (q, 5-Me), 30.0 (d, C-7), 34.2 (t, C-12), 34.5 (t, C-1), 39.3 (d, C-2), 51.4 (t, C-6), 86.3 (s, C-5), 125.4, 126.7, 127.9, 130.4, 136.8, 146.2 (4 d, 2 s, Ar), 181.5 (s, C-3) ppm. IR (KBr): = 3065–2825 (=C-H, C-H), 1755 (C=O) cm–1. C15H18O2 (230.3): calcd. C 78.23, H 7.88; found: C 78.49, H 7.93.
SmI2-induced cyclization of 11
General procedure A: Compound 11 (0.290 g, 1.01 mmol), SmI2 (2.22 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.150 g, 2.02 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 12 (0.157 g, 54%) as a colourless oil and 13 (0.072 g, 28%) as colourless crystals (mp 117–119 °C).
Methyl (6RS,8RS,10SR)-8-hydroxy-8-isopropyl-10-methyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (12): Compound 12 shows temperature-dependent NMR spectra. At rt most signals appear very broad; however, for measurements at 50 °C, the signals are clearly seen. 1H NMR (CDCl3, 500 MHz, 50 °C): δ = 0.85, 0.86 (2 d, J = 6.9 Hz, 2 × 3 H, CHMe2), 1.08 (dd, J = 12.5, 14.7 Hz, 1 H, 7-H), 1.23 (br. s, 1 H, OH), 1.35 (d, J = 7.0 Hz, 3 H, 10-Me), 1.55 (sept, J = 6.9 Hz, 1 H, CHMe2), 1.62–1.69, 3.07–3.13 (2 m, 3 H, 2 H, 5-H, 6-H, 7-H, 9-H), 3.45 (dqd, J = 3.1, 7.0, 9.9 Hz, 1 H, 10-H), 3.54 (dd, J = 7.6, 14.0 Hz, 1 H, 5-H), 3.71 (s, 3 H, CO2Me), 6.99–7.01, 7.09–7.12, 7.21–7.30 (3 m, 1 H, 1 H, 2 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz, 50 °C): δ = 16.7, 17.2 (2 q, CHMe2), 23.3 (q, 10-Me), 29.1 (d, C-10), 31.9 (t, C-7), 32.7 (t, C-5), 42.2 (d, C-6), 43.2 (d, CHMe2), 50.3 (t, C-9), 74.8 (s, C-8), 124.7, 125.8, 127.1, 129.7, 136.6, 146.9 (4 d, 2 s, Ar), 51.6, 176.2 (q, s, CO2Me) ppm. IR (neat): = 3515 (br, O-H), 3100–2845 (=C-H, C-H), 1720 (C=O) cm–1. MS (EI, 70 eV): m/z (%) = 290 (13) [M]+, 247 (70), 215 (85), 187 (54), 145 (73), 117 (40), 71 (26), 43 (100). C18H26O3 (290.4): calcd: C 74.45, H 9.02; found: C 74.71, H 8.83.
(2RS,5SR,7SR)-5-Isopropyl-7-methyl-1,5,6,7-tetrahydro-2,5-methano-4-benzoxonin-3(2H)-one (13): 1H NMR (CDCl3, 500 MHz): δ = 0.79, 0.89 (2 d, J = 6.9 Hz, 2 × 3 H, CHMe2), 1.27 (dd, J = 1.3, 13.9 Hz, 1 H, 12-H), 1.35 (d, J = 7.0 Hz, 3 H, 7-Me), 1.60 (dd, J = 11.1, 14.4 Hz, 1 H, 6-H), 1.75 (dd, J = 10.8, 13.9 Hz, 1 H, 12-H), 1.84 (sept, J = 6.9 Hz, 1 H, CHMe2), 1.95 (dd, J = 0.9, 14.4 Hz, 1 H, 6-H), 2.70–2.77 (m, 1 H, 7-H), 3.10–3.15 (m, 1 H, 2-H), 3.21 (dd, J = 12.8, 14.8 Hz, 1 H, 1-H), 3.32 (dd, J = 2.9, 14.8 Hz, 1 H, 1-H), 6.99–7.01, 7.14–7.17, 7.27–7.36 (3 m, 1 H, 1 H, 2 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 16.6, 17.4 (2 q, CHMe2), 22.9 (q, 7-Me), 29.6 (d, C-7), 30.5 (t, C-12), 34.6 (t, C-1), 38.5 (d, CHMe2), 38.9 (d, C-2), 45.3 (t, C-6), 91.2 (s, C-5), 125.4, 126.6, 127.9, 130.3, 136.7, 146.6 (4 d, 2 s, Ar), 181.7 (s, C-3) ppm. IR (KBr): = 3075–2850 (=C-H, C-H), 1765 (C=O) cm–1. MS (EI = 70 eV): m/z (%) = 258 (46) [M]+, 215 (72), 187 (41), 173 (50), 145 (69), 117 (37), 91 (28), 71 (32), 43 (100). C17H22O2 (258.4): calcd: C 79.03, H 8.58; found: C 79.09, H 8.41.
SmI2-induced reaction of 14
General procedure B: Compound 14 (0.276 g, 0.87 mmol), SmI2 (1.92 mmol), HMPA (2.75 mL, 15.7 mmol) and t-BuOH (0.129 g, 1.74 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 15 (0.103 g, 58%) as a colourless oil.
Methyl 3-(2-isopropenylphenyl)propanoate (15): 1H NMR (CDCl3, 500 MHz): δ = 2.07 [br s, 3 H, =C(Ar)Me], 2.60 (t, J = 8.2 Hz, 2 H, 2-H), 2.98 (t, J = 8.2 Hz, 2 H, 3-H), 3.69 (s, 3 H, CO2Me), 4.87 (br. s, 1 H, =CH2), 5.22 (br. s, 1 H, =CH2), 7.10–7.20 (m, 4 H, Ar) ppm. 13C NMR (CDCl3, 100 MHz): δ = 25.3 [q, =C(Ar)Me], 28.3 (t, C-3), 35.9 (t, C-2), 115.3 (t, =CH2), 126.3, 127.2, 128.4, 129.0, 137.0, 143.9, 145.4 [4 d, 3 s, =C(Ar)Me], 51.7, 173.6 (q, s, CO2Me) ppm. IR (neat): = 3065–2845 (=C-H, C-H), 1735 (C=O), 1640 (C=C) cm–1. MS (EI = 70 eV): m/z (%) = 204 (23) [M]+, 130 (47), 129 (100), 115 (42), 91 (29), 28 (47). HRMS (80 eV) calcd for C13H16O2: [M]+ = 204.11504; found: 204.11522.
SmI2-induced cyclization of 16
General procedure A: Compound 16 (0.170 g, 0.49 mmol), SmI2 (1.08 mmol), HMPA (1.54 mL, 8.77 mmol) and t-BuOH (0.073 g, 0.98 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 7:93 to 25:75) then HPLC (ethyl acetate/hexane 1:4) to furnish 17a (0.058 g, 37%, mp 162–165 °C) and 17b (0.035 g, 22%, mp 180–182 °C) as colourless crystals.
(2RS,5SR,7RS)-5-Isopropyl-7-phenyl-1,5,6,7-tetrahydro-2,5-methano-4-benzoxonin-3(2H)-one (17a): 1H NMR (CDCl3, 500 MHz): δ = 0.89, 0.98 (2 d, J = 6.9 Hz, 2 × 3 H, CHMe2), 1.37 (dd, J = 1.3, 14.0 Hz, 1 H, 12-H), 1.84 (ddd, J = 1.3, 11.0, 14.0 Hz, 1 H, 12-H), 1.93 (sept, J = 6.9 Hz, 1 H, CHMe2), 2.13 (dd, J = 11.6, 14.0 Hz, 1 H, 6-H), 2.57 (td, J = 1.3, 14.0 Hz, 1 H, 6-H), 3.21 (dddd, J = 1.3, 3.3, 11.0, 13.0 Hz, 1 H, 2-H), 3.34 (dd, J = 13.0, 15.3 Hz, 1 H, 1-H), 3.55 (dd, J = 3.3, 15.3 Hz, 1 H, 1-H), 4.03 (dd, J = 1.3, 11.6 Hz, 1 H, 7-H), 6.93–6.96, 7.00–7.05, 7.08–7.13, 7.18–7.22, 7.27–7.32 (5 m, 1 H, 1 H, 2 H, 3 H, 2 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 16.7, 17.5 (2 q, CHMe2), 30.6 (d, C-7), 34.9 (t, C-12), 39.0 (t, C-1), 39.1 (d, CHMe2), 40.7 (d, C-2), 41.5 (t, C-6), 90.8 (s, C-5), 126.5, 126.7, 128.0, 128.3, 128.5, 128.6, 130.3, 136.4, 145.2, 145.8 (7 d, 3 s, Ar), 181.5 (s, C-3) ppm. IR (KBr): = 3085–2840 (=C-H, C-H), 1760 (C=O) cm–1. MS (EI, 70 eV): m/z (%) = 320 (75) [M]+, 309 (15), 277 (92), 179 (49), 91 (92), 43 (100). HRMS (80 eV) calcd for C22H24O2: [M]+ = 320.17764; found: 320.17735.
(2RS,5SR,7SR)-5-Isopropyl-7-phenyl-1,5,6,7-tetrahydro-2,5-methano-4-benzoxonin-3(2H)-one (17b): 1H NMR (CDCl3, 500 MHz): δ = 0.93, 1.01 (2 d, J = 6.9 Hz, 2 × 3 H, CHMe2), 1.86 (sept, J = 6.9 Hz, 1 H, CHMe2), 2.34 (dd, J = 2.5, 14.5 Hz, 1 H, 6-H), 2.38 (dd, J = 9.5, 13.8 Hz, 1 H, 12-H), 2.44 (dd, J = 12.4, 14.5 Hz, 1 H, 6-H), 2.48 (dd, J = 1.0, 13.8 Hz, 1 H, 12-H), 3.29 (dddd, J = 1.0, 4.9, 9.5, 14.3 Hz, 1 H, 2-H), 3.30–3.36 (m, 2 H, 1-H), 4.40 (dd, J = 2.5, 12.4 Hz, 1 H, 7-H), 6.79–6.82, 7.04–7.07, 7.09–7.12, 7.23–7.29, 7.32–7.36 (5 m, 1 H, 2 H, 1 H, 3 H, 2 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 16.8, 17.3 (2 q, CHMe2), 37.0 (t, C-12), 37.9 (t, C-1), 39.5 (t, C-6), 40.3 (d, CHMe2), 42.5 (d, C-2), 42.8 (d, C-7), 90.6 (s, C-5), 126.2, 126.6, 128.0, 128.3, 128.3, 128.6, 132.0, 135.6, 144.3, 145.3 (7 d, 3 s, Ar), 176.2 (s, C-3) ppm. IR (KBr): = 3065–2875 (=C-H, C-H), 1755 (C=O) cm–1. MS (EI, 70 eV): m/z (%) = 320 (100) [M]+, 277 (97), 179 (45), 91 (91), 43 (59). C22H24O2 (320.4): calcd: C 82.46, H 7.55; found: C 81.83, H 7.59.
SmI2-induced cyclization of 18
General procedure A: Compound 18 (0.200 g, 0.77 mmol), SmI2 (1.70 mmol), HMPA (2.43 mL, 13.8 mmol) and t-BuOH (0.114 g, 1.54 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 19 (0.108 g, 54%) and 20 (0.040 g, 20%) as colourless oils.
Methyl (6RS,8SR,9SR)-8-hydroxy-8,9-dimethyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (19): Compound 19 shows temperature-dependent NMR spectra. At rt several signals appear broad; however, for measurements at 55 °C, signals are more clearly seen. 1H NMR (CDCl3, 500 MHz, 55 °C): δ = 0.97 (d, J = 7.0 Hz, 3 H, 9-Me), 1.17 (s, 3 H, 8-Me), 1.62 (dd, J = 2.8, 14.8 Hz, 1 H, 7-H), 1.77 (dd, J = 12.0, 14.8 Hz, 1 H, 7-H), 1.94–2.01 (m, 1 H, 9-H), 2.54 (dd, J = 9.1, 14.0 Hz, 1 H, 10-H), 2.96–3.08 (m, 3 H, 5-H, 6-H, 10-H), 3.22 (dd, J = 5.0, 13.8 Hz, 1 H, 5-H), 3.70 (s, 3 H, CO2Me), 7.07–7.15 (m, 4 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz, 55 °C): δ = 17.0 (q, 9-Me), 32.0 (q, 8-Me), 35.1 (t, C-5), 35.8 (t, C-7), 37.4 (t, C-10), 43.0 (d, C-6), 47.0 (d, C-9), 74.8 (s, C-8), 126.8, 127.0, 130.6, 130.7, 138.1, 139.7 (4 d, 2 s, Ar), 51.7, 176.3 (q, s, CO2Me) ppm. IR (neat): = 3510 (br, O-H), 3100–2850 (=C-H, C-H), 1720 (C=O) cm−1. C16H22O3 (262.3): calcd: C 73.25, H 8.45; found: C 73.43, H 8.30.
Methyl (2SR,4RS,4aSR)-4-hydroxy-4-methyl-8-[(1E)-propen-1-yl]-1,2,3,4,4a,7-hexahydro-naphthalene-2-carboxylate (20): 1H NMR (CDCl3, 500 MHz): δ = 1.02 (s, 3 H, 4-Me), 1.79 (dd, J = 1.5, 6.6 Hz, 3 H, =CHMe), 1.87-1.91 (m, 2 H, 1-H, 3-H), 2.07 (dd, J = 3.8, 12.5 Hz, 1 H, 3-H), 2.37 (tt, J = 3.8, 12.9 Hz, 1 H, 2-H), 2.70–2.84 (m, 3 H, 1-H, 7-H), 3.21 (dd, J = 1.6, 3.8 Hz, 1 H, 4a-H), 3.72 (s, 3 H, CO2Me), 5.64 (qd, J = 6.6, 15.6 Hz, 1 H, =CHMe), 5.84–5.87, 5.89–5.92 (2 m, 2 × 1 H, 5-H, 6-H), 6.53 (qd, J = 1.5, 15.6 Hz, 1 H, CH=CHMe) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz): δ = 18.9 (q, =CHMe), 22.0 (q, 4-Me), 27.5 (t, C-1), 31.2 (t, C-7), 41.0 (d, C-2), 44.0 (t, C-3), 50.2 (d, C-4a), 74.6 (s, C-4), 123.2, 124.8, 125.8, 126.6, 128.2, 129.3 (4 d, 2 s, =CH, =Cq), 52.0, 175.2 (q, s, CO2Me) ppm. Due to instability of the sample and storage before the MS analysis was accomplished, only the rearomatized product could be detected. HRMS (ESI) calcd for C16H20O3: [M+Na]+ = 283.1305; found: 283.1295.
SmI2-induced cyclization of 21
General procedure B: Compound 21 (0.270 g, 0.99 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9), then HPLC (iso-propanol/hexane 2:98) to furnish 22 (0.152 g, 56%) and 23 (0.012 g, 6%) as colourless oils.
Methyl (6RS,8SR,9SR)-8-ethyl-8-hydroxy-9-methyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (22): Compound 22 shows temperature-dependent NMR spectra. At rt several signals appear broad; however, for measurements at 40 °C, the signals are more clearly seen. 1H NMR (CDCl3, 500 MHz, 40 °C): δ = 0.87 (t, J = 7.2 Hz, 3 H, 8-CH2Me), 0.92 (d, J = 6.9 Hz, 3 H, 9-Me), 1.39, 1.50 (2 qd, J = 7.2, 14.4 Hz, 2 × 1 H, 8-CH2Me), 1.56–1.70 (m, 2 H, 7-H), 1.97–2.03 (m, 1 H, 9-H), 2.55 (dd, J = 8.5, 13.6 Hz, 1 H, 10-H), 2.97–3.02 (m, 1 H, 6-H), 3.05–3.13 (m, 2 H, 5-H, 10-H), 3.27 (dd, J = 4.0, 13.7 Hz, 1 H, 5-H), 3.72 (s, 3 H, CO2Me), 7.08–7.11, 7.14–7.18 (2 m, 1 H, 3 H, Ar) ppm, the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz, 40 °C): δ = 7.1 (q, 8-CH2Me), 16.0 (q, 9-Me), 33.4 (t, C-7), 35.3 (t, C-5), 36.6 (t, 8-CH2Me), 36.8 (t, C-10), 43.0 (d, C-6), 44.2 (d, C-9), 75.7 (s, C-8), 126.7, 126.9, 130.6, 130.7, 138.3, 139.7 (4 d, 2 s, Ar), 51.6, 176.4 (q, s, CO2Me) ppm. IR (neat): = 3535 (br, O-H), 3060–2860 (=C-H, C-H), 1720 (C=O) cm–1. HRMS (ESI) calcd for C17H24O3: [M+Na]+ = 299.1618; found: 299.1614.
Methyl 3-{2-[(1E)-propen-1-yl]phenyl}propanoate (23): 1H NMR (CDCl3, 400 MHz): δ = 1.90 (dd, J = 1.6, 6.6 Hz, 3 H, =CHMe), 2.57 (t, J = 8.2 Hz, 2 H, 2-H), 2.99 (t, J = 8.2 Hz, 2 H, 3-H), 3.69 (s, 3 H, CO2Me), 6.12 (qd, J = 6.6, 15.5 Hz, 1 H, =CHMe), 6.62 (qd, J = 1.6, 15.5 Hz, 1 H, =CHAr), 7.10–7.21, 7.39–7.42 (2 m, 3 H, 1 H, Ar) ppm. 13C NMR (CDCl3, 100 MHz): δ = 19.0 (q, =CHMe), 28.6 (t, C-3), 35.2 (t, C-2), 126.2, 126.8, 127.2, 128.1, 128.2, 129.3, 136.9, 137.2 (6 d, 2 s, ArCH=CH), 51.8, 173.6 (q, s, CO2Me) ppm. IR (neat): = 3065–2850 (=C-H, C-H), 1735 (C=O), 1600 (C=C) cm–1. HRMS (ESI) calcd for C13H16O2: [M+Na]+ = 227.1043, [M+K]+ = 243.0782; found: 227.1018, 243.0936.
SmI2-induced cyclization of 24
General procedure A: Compound 24 (0.300 g, 1.04 mmol), SmI2 (2.30 mmol), HMPA (3.30 mL, 18.8 mmol) and t-BuOH (0.156 g, 2.10 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 25 (0.188 g, 63%) as colourless crystals (mp 89–90 °C).
Methyl (6RS,8RS,9SR)-8-hydroxy-8-isopropyl-9-methyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (25): Compound 25 shows temperature-dependent spectra. At rt most signals appear very broad; however, for measurements at 65 °C, the signals are clearly seen; 1H NMR (d6-DMSO, 500 MHz, 65 °C): δ = 0.78 (d, J = 7.0 Hz, 3 H, 9-Me), 0.85, 0.86 (2 d, J = 6.7 Hz, 2 × 3 H, CHMe2), 1.14 (dd, J = 12.7, 14.9 Hz, 1 H, 7-H), 1.40 (dd, J = 2.7, 14.9 Hz, 1 H, 7-H), 1.49 (sept, J = 6.7 Hz, 1 H, CHMe2), 2.01–2.07 (m, 1 H, 9-H), 2.33 (dd, J = 5.9, 13.8 Hz, 1 H, 10-H), 2.92 (dd, J = 2.4, 13.4 Hz, 1 H, 5-H), 2.99 (dddd, J = 2.4, 2.7, 6.6, 12.7 Hz, 1 H, 6-H), 3.33 (dd, J = 2.0, 13.8 Hz, 1 H, 10-H), 3.44 (dd, J = 6.6, 13.4 Hz, 1 H, 5-H), 3.61 (s, 3 H, CO2Me), 6.89–6.91, 7.06–7.12 (2 m, 1 H, 3 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (d6-DMSO, 125 MHz, 65 °C): δ = 13.3 (q, 9-Me), 15.7, 16.2 (2 q, CHMe2), 28.0 (t, C-7), 33.2 (t, C-5), 34.9 (t, C-10), 36.0 (d, CHMe2), 39.9 (d, C-9), 40.9 (d, C-6), 74.8 (s, C-8), 125.2, 125.7, 129.5, 130.8, 137.4, 139.3 (4 d, 2 s, Ar), 50.8, 175.1 (q, s, CO2Me) ppm. IR (KBr): = 3490 (br, O-H), 3000–2840 (=C-H, C-H), 1715 (C=O) cm–1. MS (EI = 70 eV): m/z (%) = 290 (24) [M]+, 247 (74), 215 (90), 187 (100), 157 (54), 117 (54), 71 (21), 43 (80). C18H26O3 (290.4): calcd: C 74.45, H 9.02; found: C 74.51, H 8.74.
SmI2-induced cyclization of 26a
General procedure A: Compound 26a (0.300 g, 1.00 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9), then HPLC (ethyl acetate/hexane 15:85) to furnish 27 and 28 (0.054 g, 18%) as an inseparable mixture (27:28 = 5:3 by integration of NMR signals) and 26a (0.125 g, 42%) as colourless oils.
Methyl (4aSR,5RS,12SR,12aRS)-12a-hydroxy-12-methyl-1,2,3,4,4a,5,6,11,12,-12a-decahydro-dibenzo[a,e][8]annulene-5-carboxylate (27): 1H NMR (CDCl3, 700 MHz): δ = 0.80–2.03 (m, 10 H, 1-H, 2-H, 3-H, 4-H, 4a-H, OH), 1.04 (d, J = 6.6 Hz, 3 H, 12-Me), 2.56–2.62 (m, 1 H, 12-H), 2.70 (dd, J = 11.3, 16.8 Hz, 1 H, 11-H), 2.82 (dd, J = 2.2, 15.0 Hz, 1 H, 6-H), 2.97 (dd, J = 3.8, 16.8 Hz, 1 H, 11-H), 3.00 (ddd, J = 2.2, 7.3, 9.6 Hz, 1 H, 5-H), 3.61 (dd, J = 7.3, 15.0 Hz, 1 H, 6-H), 3.63 (s, 3 H, CO2Me), 7.03–7.07, 7.10–7.15 (2 m, 2 × 2 H, Ar) ppm.
Methyl (4aS,4bR,8aR,9S)-4b-hydroxy-1-[(1E)-prop-1-en-1-yl]-2,4a,4b,5,6,7,8,8a,-9,10-decahydro-phenanthrene-9-carboxylate (28): 1H NMR (CDCl3, 700 MHz): δ = 0.80–2.03 (m, 15 H, 2-H, 5-H, 6-H, 7-H, 8-H, 8a-H, 9-H, 10-H, OH), 1.81 (dd, J = 1.2, 6.5 Hz, 3 H, =CHMe), 3.14 (dd, J = 3.8, 13.3 Hz, 1 H, 4a-H), 3.72 (s, 3 H, CO2Me), 5.66 (qd, J = 6.5, 15.4 Hz, 1 H, =CHMe), 5.83–5.85, 5.90–5.93 (2 m, 2 × 1 H, 3-H, 4-H), 6.53 (br. d, J = 15.4 Hz, 1 H, 1-CH=) ppm.
Mixture of 27 + 28: 13C NMR (CDCl3, 175 MHz, 40 °C): δ = 17.3, 18.9, 20.1, 20.7, 21.2, 24.4, 25.2, 26.6, 27.4, 27.6, 30.5, 32.0, 37.3, 40.2, 41.5, 43.0, 44.2, 45.2, 45.3, 51.2, 51.5, 51.9, 75.4 (s, C-4b, 28), 75.9 (s, C-12a, 27), 123.0, 124.7, 126.0, 126.2, 128.0, 129.2 (4 d, 2 s, C-1, C-3, C-4, C-10a, CH=CHMe, 28), 125.8, 126.7, 129.5, 132.6, 136.0, 140.4 (4 d, 2 s, Ar, 27), 175.5 (s, C=O, 28), 176.6 (s, C=O, 27) ppm, no unambiguous assignment of signals was possible. IR (neat): = 3510 (br, OH), 3060–2860 (=C-H, C-H), 1720 (br, C=O), 1600 (C=C) cm–1. HRMS (ESI) calcd for C19H26O3: [M+Na]+ = 325.1774; found: 325.1798.
SmI2-induced cyclization of 26b
General procedure A: Compound 26b (0.300 g, 1.00 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 29 (0.025 g, 9%) as colourless crystals (mp 119–121 °C), 23 (0.082 g, 27%) and 26b (0.120 g, 40%) as colourless oils.
(4aRS,5RS,12RS,12aSR)-5-Methyl-1,3,4,5,6,11,12,12a-octahydro-2H-4a,12-(epoxymethano)dibenzo[a,e][8]annulen-13-one (29): 1H NMR (CDCl3, 500 MHz): δ = 0.90 (d, J = 7.1 Hz, 3 H, 5-Me), 1.25–1.31, 1.39–1.48, 1.52–1.60, 1.65–1.81 (4 m, 1 H, 2 × 2 H, 3 H, 1-H, 2-H, 3-H, 4-H), 1.97–2.12 (m, 1 H, 12a-H), 2.19 (dqd, J = 2.0, 7.1, 7.2 Hz, 1 H, 12a-H), 2.57 (dd, J = 7.2, 14.6 Hz, 1 H, 6-H), 2.79 (br. d, J ≈ 14.6 Hz, 1 H, 6-H), 2.82 (ddd, J = 1.0, 5.2, 9.6 Hz, 1 H, 12-H), AB part of ABX system (δA = 3.24, δB = 3.27, JAB = 15.2 Hz, JAX = 9.6 Hz, JBX = 5.2 Hz, 2 H, 11-H), 7.05–7.07, 7.14–7.21 (2 m, 1 H, 3 H, Ar) ppm. 13C NMR (CDCl3, 100 MHz): δ = 15.4 (q, 5-Me), 16.4, 16.5 (2 t, C-2, C-3), 28.6, 28.8 (2 t, C-1, C-4), 35.0 (t, C-6), 35.8 (t, C-11), 42.0 (d, C-5), 47.4 (d, C-12), 91.3 (s, C-4a), 126.7, 126.9, 131.3, 132.1, 137.7, 142.6 (4 d, 2 s, Ar) ppm, the signal for C-13 is not clearly visible. Signals for C-2, C-3, C-5, C-6, C-11, C-12, 5-Me, and three of the aromatic carbon atoms are broad. IR (KBr): = 3045–2870 (=C-H, C-H), 1745 (C=O) cm–1. HRMS (ESI) calcd for C18H22O2: [M+H]+ = 271.1693, [M+Na]+ = 293.1512; found: 271.1692, 293.1509.
SmI2-induced cyclization of 30
General procedure B: Compound 30 (0.288 g, 1.00 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 31 (0.022 g, 8%) as colourless crystals (mp 81–83 °C), 32 (0.064 g, 31%) and 30 (0.092 g, 32%) as colourless oils.
Methyl (6RS,8RS,9RS)-8-hydroxy-8-isopropyl-9-methyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (31): Compound 31 shows temperature-dependent spectra. At rt most signals appear very broad; for measurements at 55 °C, the signals are clearly seen. 1H NMR (CDCl3, 500 MHz, 55 °C): δ = 0.89, 0.90 (2 d, J = 6.8 Hz, 2 × 3 H, CHMe2), 0.94 (d, J = 7.0 Hz, 3 H, 9-Me), 1.49–1.65 (m, 1 H, 7-H), 1.62 (sept, J = 6.8 Hz, 1 H, CHMe2), 1.71–1.82 (m, 1 H, 7-H), 2.04–2.11 (m, 1 H, 9-H), 2.57 (dd, J = 8.4, 13.2 Hz, 1 H, 10-H), 2.87–2.97 (m, 1 H, 6-H), 3.07 (dd, J = 5.9, 13.7 Hz, 1 H, 5-H), 3.03–3.14 (m, 1 H, 10-H), 3.24–3.35 (m, 1 H, 5-H), 3.72 (s, 3 H, CO2Me), 7.07–7.09, 7.13–7.18 (2 m, 1 H, 3 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz, 55 °C): δ = 15.4 (q, 9-Me), 16.3, 16.5 (2 q, CHMe2), 36.7 (d, CHMe2), 42.8 (d, C-9), 65.9 (s, C-8), 126.7, 127.0, 130.7, 130.8, 138.6, 139.9 (4 d, 2 s, Ar), 51.7, 176.5 (q, s, CO2Me) ppm. The signals for C-5, C-6 and C-7 are not clearly visible, the signals for C-9 and 9-Me are broad. IR (KBr): = 3490 (br, O-H), 3060–2850 (=C-H, C-H), 1710 (C=O) cm–1. HRMS (ESI) calcd for C18H26O3: [M+Na]+ = 313.1774; found: 313.1773.
Methyl 3-{2-[(1Z)-propen-1-yl]phenyl}propanoate (32): 1H NMR (CDCl3, 250 MHz): δ = 1.72 (dd, J = 1.6, 7.0 Hz, 3 H, =CHMe), 2.54 (t, J = 8.2 Hz, 2 H, 2-H), 2.92 (t, J = 8.2 Hz, 2 H, 3-H), 3.66 (s, 3 H, CO2Me), 5.85 (qd, J = 7.0, 11.5 Hz, 1 H, =CHMe), 6.52 (qd, J = 1.6, 11.5 Hz, 1 H, =CHAr), 7.18 (br. s, 4 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 14.4 (q, =CHMe), 28.8 (t, C-3), 34.8 (t, C-2), 126.1, 127.1, 127.8, 128.4, 129.0, 129.8, 136.4, 138.7 (6 d, 2 s, ArCH=CH), 51.7, 173.6 (q, s, CO2Me) ppm. IR (neat): = 3065–2870 (=C-H, C-H), 1735 (C=O) cm–1. MS (EI, 70 eV): m/z (%) = 204 (56) [M]+, 144 (36), 129 (100), 115 (88), 91 (51), 77 (19), 18 (70); HRMS (80 eV) calcd for C13H16O2: [M]+ = 204.11504; found: 204.11464.
SmI2-induced cyclization of 33
General procedure B: Compound 33 (0.314 g, 1.00 mmol), SmI2 (2.20 mmol), HMPA (3.16 mL, 18.0 mmol) and t-BuOH (0.148 g, 2.00 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 34 (0.075 g, 24%) as colourless crystals (mp 131–133 °C) and 35 (0.109 g, 47%) as a colourless oil.
Methyl (6RS,8RS,9RS)-9-cyclopropyl-8-hydroxy-8-isopropyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (34): Compound 35 shows temperature-dependent spectra. At rt most signals appear very broad; however, for measurements at 51 °C, the signals are clearly seen. 1H NMR (CDCl3, 500 MHz, 51 °C): δ = 0.27–0.32, 0.47–0.57, 0.62–0.69 (3 m, 1 H, 3 H, 1 H, 1'-H, 2'-H, 3'-H), 0.90, 0.98 (2 d, J = 6.7 Hz, 2 × 3 H, CHMe2), 1.12 (s, 1 H, OH), 1.28–1.33 (m, 1 H, 9-H), 1.60–1.68, 1.87–1.91 (2 m, 2 × 1 H, 7-H), 2.06 (sept, J = 6.7 Hz, 1 H, CHMe2), 2.68 (dd, J = 6.9, 13.9 Hz, 1 H, 10-H), 2.94–2.99 (m, 1 H, 6-H), 3.07 (dd, J = 4.1, 13.9 Hz, 1 H, 10-H), 3.18–3.21 (m, 1 H, 5-H), 3.41 (dd, J = 5.6, 13.7 Hz, 1 H, 5-H), 3.71 (s, 3 H, CO2Me), 7.04–7.07, 7.11–7.15, 7.26–7.30 (3 m, 1 H, 2 H, 1 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz, 51 °C): δ = 3.8, 6.6 (2 t, C-2', C-3'), 11.7 (d, C-1'), 17.1, 17.3 (2 q, CHMe2), 34.8, 35.2, 35.9 (d, 2 t, CHMe2, C-5, C-10), 41.9 (d, C-6), 51.4 (d, C-9), 77.7 (s, C-8), 126.2, 126.5, 130.3, 131.0, 138.1, 140.2 (4 d, 2 s, Ar), 51.8, 176.5 (q, s, CO2Me) ppm, the signal for C-7 is not clearly visible, the signals for C-5, C-8, C-9, C-10, C-1', C-2', C-3', CHMe2, Ar (2 d, 2 s) are broad. IR (KBr): = 3490 (br, O-H), 3080–2875 (=C-H, C-H), 1715 (C=O) cm–1. C20H28O3 (316.4): calcd: C 75.91, H 8.92; found: C 76.09, H 8.88.
Methyl 3-{2-[(E)-2-cyclopropylvinyl]phenyl}propanoate (35): 1H NMR (CDCl3, 500 MHz): δ = 0.82–0.91 (m, 4 H, 2'-H, 3'-H), 1.61 (ttd, J = 4.5, 8.5, 8.9 Hz, 1 H, 1'-H), 2.59 (t, J = 8.2 Hz, 2 H, 2-H), 3.02 (t, J = 8.2 Hz, 2 H, 3-H), 3.70 (s, 3 H, CO2Me), 5.60 (dd, J = 8.9, 15.5 Hz, 1 H, 1'-CH=), 6.69 (d, J = 15.5 Hz, 1 H, =CHAr), 7.12–7.18, 7.36–7.39 (2 m, 3 H, 1 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 7.5 (t, C-2', C-3'), 15.0 (d, C-1'), 28.6 (t, C-2), 35.2 (t, C-3), 124.4 (d, =CHAr), 137.2 (d, 1'-CH=), 125.8, 126.8, 126.9, 129.3, 136.6, 137.0 (4 d, 2 s, Ar), 51.8, 173.6 (q, s, CO2Me) ppm. HRMS (ESI) calcd for C15H18O2: [M+Na]+ = 253.1199; found: 253.1201.
SmI2-induced cyclization of 36
General procedure B: Compound 36 (0.200 g, 0.57 mmol), SmI2 (1.26 mmol), HMPA (1.80 mL, 10.3 mmol) and t-BuOH (0.085 g, 1.15 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 37 (0.094 g, 47%) as colourless crystals (mp 135–137 °C) and 36 (0.040 g, 20%) as a colourless oil.
Methyl (6RS,8RS,9RS)-8-hydroxy-8-isopropyl-9-phenyl-5,6,7,8,9,10-hexahydro-benzo[8]annulene-6-carboxylate (37): 1H NMR (CDCl3, 500 MHz): δ = 1.07 (br. s, 1 H, OH), 1.10, 1.17 (2 d, J = 6.8 Hz, 2 × 3 H, CHMe2), 2.06 (dd, J = 12.4, 14.2 Hz, 1 H, 7-H), 2.07 (sept, J = 6.8 Hz, 1 H, CHMe2), 2.40–2.44 (m, 1 H, 7-H), 2.81 (dddd, J = 1.8, 2.6, 12.3, 12.4 Hz, 1 H, 6-H), 2.91 (dd, J = 3.0, 13.1 Hz, 1 H, 10-H), 3.02 (dd, J = 1.8, 14.8 Hz, 1 H, 5-H), 3.08 (dd, J = 11.9, 13.1 Hz, 1 H, 10-H), 3.21 (dd, J = 3.0, 11.9 Hz, 1 H, 9-H), 3.44 (dd, J = 12.3, 14.8 Hz, 1 H, 5-H), 3.74 (s, 3 H, CO2Me), 6.52–6.54, 6.75–6.77, 6.85–6.88, 7.04–7.14 (4 m, 1 H, 2 H, 1 H, 5 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 17.5, 17.7 (2 q, CHMe2), 35.2 (t, C-10), 35.3 (d, CHMe2), 38.3 (t, C-5), 38.5 (t, C-7), 40.5 (d, C-6), 57.8 (d, C-9), 74.2 (s, C-8), 125.9, 126.5, 127.5, 128.1, 128.9, 130.9, 134.4, 137.6, 139.4, 140.5 (7 d, 3 s, Ar), 52.1, 176.8 (q, s, CO2Me) ppm. IR (KBr): = 3485 (br, O-H), 3100–2845 (=C-H, C-H), 1710 (C=O) cm–1. MS (EI = 70 eV): m/z (%) = 352 (41) [M]+, 309 (15), 267 (22), 157 (70), 91 (100), 71 (40), 43 (95). C23H28O3 (352.5): calcd C 78.38, H 8.01; found: C 78.05, H 8.04.
SmI2-induced cyclization of 38a
General procedure A: Compound 38a (0.170 g, 0.47 mmol), SmI2 (1.04 mmol), HMPA (1.48 mL, 8.43 mmol) and t-BuOH (0.070 g, 0.94 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9), then HPLC (ethyl acetate/hexane 15:85) to furnish 39 (0.074 g, 44%) as colourless crystals (mp 114–115 °C).
Methyl (4aSR,5SR,11RS,11aSR)-5-benzyl-4a-hydroxy-2,3,4,4a,5,10,11,11a-octahydro-1H-dibenzo[a,d][7]annulene-11-carboxylate (39): 1H NMR (CDCl3, 500 MHz): δ = 1.10 (s, 1 H, OH), 1.24–1.36, 1.42–1.51, 1.59–1.64, 1.66–1.75, 2.01–2.08 (5 m, 2 H, 2 × 1 H, 3 H, 1 H, 1-H, 2-H, 3-H, 4-H), 2.24 (ddd, J = 3.7, 11.2, 11.4 Hz, 1 H, 11a-H), 2.56 (ddd, J = 1.6, 11.2, 12.2 Hz, 1 H, 11-H), 2.75 (dd, J = 1.6, 15.0 Hz, 1 H, 10-H), 2.81 (dd, J = 3.3, 11.2 Hz, 1 H, 5-H), AB part of ABX system (δA = 3.11, δB = 3.19, JAB = 13.3 Hz, JAX = 3.3 Hz, JBX = 11.2 Hz, 2 H, PhCH2), 3.57 (dd, J = 12.2, 15.0 Hz, 1 H, 10-H), 3.73 (s, 3 H, CO2Me), 6.54–6.56, 6.81–6.83, 6.87–6.91, 7.05–7.12 (4 m, 1 H, 2 H, 1 H, 5 H, Ar) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz): δ = 21.9 (t, C-3), 25.8 (t, C-2), 28.5 (t, C-1), 35.7 (t, PhCH2), 38.6 (t, C-10), 39.7 (t, C-4), 44.7 (d, C-11a), 47.8 (d, C-11), 64.9 (d, C-5), 72.7 (s, C-4a), 125.9, 126.7, 127.5, 128.1, 128.9, 130.6, 133.7, 138.1, 138.6, 141.0 (7 d, 3 s, Ar), 51.7, 176.6 (q, s, CO2Me) ppm. IR (KBr): = 3430 (br, O-H), 3055–2845 (=C-H, C-H), 1705 (C=O) cm–1. MS (EI = 70 eV): m/z (%) = 364 (4) [M]+, 267 (39), 169 (39), 115 (23), 91 (100), 41 (10). HRMS (80 eV) calcd for C24H28O3: [M]+ = 364.2039; found: 364.2034.
SmI2-induced cyclization of 38b
General procedure A: Compound 38b (0.145 g, 0.40 mmol), SmI2 (0.88 mmol), HMPA (1.27 mL, 7.23 mmol) and t-BuOH (0.059 g, 0.80 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9) to furnish 40 (0.084 g, 63%) as colourless crystals (mp 193–195 °C).
(4aRS,5RS,11RS,11aRS)-5-Benzyl-1,2,3,4,5,10,11,11a-octahydro-4a,11-(epoxy-methano)-dibenzo[a,d][7]annulen-12-one (40): 1H NMR (CDCl3, 500 MHz): δ = 1.11–1.29, 1.47–1.56, 1.71–1.75, 1.95–2.03, 2.19–2.23 (5 m, 2 H, 1 H, 2 × 2 H, 1 H, 1-H, 2-H, 3-H, 4-H), 2.54 (dd, J = 6.6, 11.4 Hz, 1 H, 11a-H), 2.65 (dd, J = 11.3, 12.8 Hz, 1 H, PhCH2), 2.71 (dd, J = 2.6, 6.6 Hz, 1 H, 11-H), 2.94 (dd, J = 2.4, 12.8 Hz, 1 H, PhCH2), 3.02 (dd, J = 2.4, 11.3 Hz, 1 H, 5-H), 3.23–3.31 (m, 2 H, 10-H), 6.29–6.31, 6.71–6.74, 6.81–6.85, 7.04–7.07, 7.11–7.15 (5 m, 1 H, 2 H, 2 × 1 H, 4 H, Ar) ppm. 13C NMR (CDCl3, 125 MHz): δ = 21.5 (t, C-3), 23.6 (t, C-2), 29.9 (t, C-1), 33.7 (t, C-4), 36.6 (t, C-10), 39.6 (t, PhCH2), 43.5 (d, C-11a), 49.5 (d, C-5), 62.4 (d, C-11), 86.0 (s, C-4a), 126.3, 126.5, 127.1, 128.2, 129.2, 131.2, 133.3, 135.5, 137.6, 139.6 (7 d, 3 s, Ar), 178.2 (s, C-12) ppm. IR (neat): = 3060–2855 (=C-H, C-H), 1760 (C=O) cm–1. C23H24O2 (332.4): calcd: C 83.10, H 7.28; found: C 82.64, H 7.08.
SmI2-induced cyclization of 41
General procedure A: Compound 41 (0.223 g, 0.81 mmol), SmI2 (1.80 mmol), HMPA (2.56 mL, 14.6 mmol) and t-BuOH (0.120 g, 1.62 mmol). The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane 1:9), then HPLC (ethyl acetate/hexane 1:5) to furnish 42 (0.031 g, 14%) and 43 (0.014 g, 7%) as colourless oils.
Methyl (2SR,4RS,4aSR)-4-hydroxy-4-methyl-8-(2-methylpropen-1-yl)-1,2,3,4,4a,7-hexahydro-naphthalene-2-carboxylate (42): 1H NMR (CDCl3, 500 MHz): δ = 1.08 (s, 3 H, 4-Me), 1.54, 1.74 (2 s, 2 × 3 H, =CMe2), 1.75–1.82 (m, 2 H, 1-H, 3-H), 2.04 (ddd, J = 1.8, 3.8, 12.5 Hz, 1 H, 3-H), 2.38 (tt, J = 3.8, 12.9 Hz, 1 H, 2-H), 2.50–2.62 (m, 2 H, 7-H), 2.71–2.77 (m, 2 H, 1-H, 4a-H), 3.67 (s, 3 H, CO2Me), 5.50 (br. s, 1 H, CH=CMe2), 5.82–5.88 (m, 2 H, 5-H, 6-H) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz): δ = 19.5, 25.3 (2 q, =CMe2), 22.3 (q, 4-Me), 31.5 (t, C-7), 32.6 (t, C-1), 40.5 (d, C-2), 44.0 (t, C-3), 49.3 (d, C-4a), 74.2 (s, C-4), 123.7, 124.7, 125.9, 127.5, 128.8, 134.2 (3 d, 3 s, =CH, =Cq), 51.9, 175.5 (q, s, CO2Me) ppm.
(1SR,4SR,9aSR)-1-Methyl-6-(2-methylpropen-1-yl)-4,5,7,9a-tetrahydro-1,4-methano-2-benzoxepin-3(1H)-one (43): 1H NMR (CDCl3, 500 MHz): δ = 1.51, 1.76 (2 s, 2 × 3 H, =CMe2), 1.54 (s, 3 H, 4-Me), AB part of ABX system (δA = 1.86, δB = 1.91, JAB = 11.5 Hz, JAX = 4.7 Hz, JBX too small to see signal splitting, 2 H, 10-H), 2.22–2.27 (m, 1 H, 5-H), 2.52–2.67 (m, 3 H, 5-H, 7-H), 2.84–2.90 (m, 1 H, 4-H), 3.16–3.20 (m, 1 H, 9a-H), 5.49 (br. s, 1 H, CH=CMe2), 5.74–5.78, 5.84–5.88 (2 m, 2 × 1 H, 8-H, 9-H) ppm, the signal for the OH group could not be assigned unambiguously. 13C NMR (CDCl3, 125 MHz): δ = 19.7, 25.4 (2 q, =CMe2), 23.7 (q, 1-Me), 30.1 (t, C-5), 31.6 (t, C-7), 37.1, 37.9 (2 d, C-4, C-9a), 45.3 (t, C-10), 88.7 (s, C-1), 123.9, 124.2, 124.4, 126.8, 131.9, 136.1 (3 d, 3 s, =CH, =Cq), 180.9 (s, C-3) ppm.
Supporting Information
| Supporting Information File 1:
Experimental procedures and characterization data of synthesized compounds.
Supporting Information File 1 contains all experimental procedures for the syntheses of the starting materials 2, 4, 5, 7, 11, 14, 16, 18, 21, 24, 26, 30, 33, 36, 38, and 41 and their analytical data. |
||
| Format: PDF | Size: 210.8 KB | Download |
References
-
Mehta, G.; Singh, V. Chem. Rev. 1999, 99, 881–930. doi:10.1021/cr9800356
Return to citation in text: [1] [2] -
Petasis, N. A.; Patane, M. A. Tetrahedron 1992, 48, 5757–5821. doi:10.1016/S0040-4020(01)90172-3
Return to citation in text: [1] [2] -
Galli, C.; Mandolini, L. Eur. J. Org. Chem. 2000, 3117–3125. doi:10.1002/1099-0690(200009)2000:18<3117::AID-EJOC3117>3.0.CO;2-5
Return to citation in text: [1] -
Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. doi:10.1002/anie.200500369
Return to citation in text: [1] [2] -
Fujiwara, K.; Goto, A.; Sato, D.; Kawai, H.; Suzuki, T. Tetrahedron Lett. 2005, 46, 3465–3468. doi:10.1016/j.tetlet.2005.03.114
Return to citation in text: [1] [2] -
Nair, V.; Abhilash, K. G. Synlett 2008, 301–312. doi:10.1055/s-2008-1032058
Return to citation in text: [1] [2] -
Ohno, H.; Hamaguchi, H.; Ohata, M.; Kosaka, S.; Tanaka, T. J. Am. Chem. Soc. 2004, 126, 8744–8754. doi:10.1021/ja048693x
Return to citation in text: [1] [2] -
Yu, Z.-X.; Wang, Y.; Wang, Y. Chem.–Asian J. 2010, 5, 1072–1088. doi:10.1002/asia.200900712
Return to citation in text: [1] [2] -
Genrich, F.; Schaumann, E. Tetrahedron Lett. 2009, 50, 6187–6190. doi:10.1016/j.tetlet.2009.08.092
Return to citation in text: [1] [2] -
Zhang, T.; Huang, X.; Xue, J.; Sun, S. Tetrahedron Lett. 2009, 50, 1290–1294. doi:10.1016/j.tetlet.2009.01.001
Return to citation in text: [1] [2] -
Evans, P. A.; Holmes, B. Tetrahedron 1991, 47, 9131–9166. doi:10.1016/S0040-4020(01)96203-9
Return to citation in text: [1] -
Roxburgh, C. J. Tetrahedron 1993, 49, 10749–10784. doi:10.1016/S0040-4020(01)80232-5
Return to citation in text: [1] -
Rousseau, G. Tetrahedron 1995, 51, 2777–2849. doi:10.1016/0040-4020(94)01064-7
Return to citation in text: [1] -
Hoberg, J. O. Tetrahedron 1998, 54, 12631–12670. doi:10.1016/S0040-4020(98)00596-1
Return to citation in text: [1] -
Yet, L. Tetrahedron 1999, 55, 9349–9403. doi:10.1016/S0040-4020(99)00483-4
Return to citation in text: [1] -
Yet, L. Chem. Rev. 2000, 100, 2963–3008. doi:10.1021/cr990407q
Return to citation in text: [1] -
Nubbemeyer, U. Top. Curr. Chem. 2001, 216, 125–196. doi:10.1007/3-540-44726-1_4
Return to citation in text: [1] -
Shiina, I. Chem. Rev. 2007, 107, 239–273. doi:10.1021/cr050045o
Return to citation in text: [1] -
Molander, G. A. Acc. Chem. Res. 1998, 31, 603–609. doi:10.1021/ar960101v
Return to citation in text: [1] -
Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429
Return to citation in text: [1] [2] [3] [4] -
Procter, D. J.; Flowers, R. A., II; Skrydstrup, T. Organic Synthesis using Samarium Diiodide; Royal Society of Chemistry: Cambridge, U.K., 2009.
Return to citation in text: [1] -
Molander, G. A.; McKie, J. A. J. Org. Chem. 1994, 59, 3186–3192. doi:10.1021/jo00090a041
Return to citation in text: [1] -
Molander, G. A.; Alonso-Alija, C. J. Org. Chem. 1998, 63, 4366–4373. doi:10.1021/jo980119e
Return to citation in text: [1] -
Molander, G. A.; Machrouhi, F. J. Org. Chem. 1999, 64, 4119–4123. doi:10.1021/jo990216n
Return to citation in text: [1] -
Molander, G. A.; Köllner, C. J. Org. Chem. 2000, 65, 8333–8339. doi:10.1021/jo001195w
Return to citation in text: [1] -
Molander, G. A.; Le Huérou, Y.; Brown, G. A. J. Org. Chem. 2001, 66, 4511–4516. doi:10.1021/jo001513r
Return to citation in text: [1] -
Molander, G. A.; Brown, G. A.; Storch de Gracia, I. J. Org. Chem. 2002, 67, 3459–3463. doi:10.1021/jo020027w
Return to citation in text: [1] -
Inanaga, J.; Yokoyama, Y.; Handa, Y.; Yamaguchi, M. Tetrahedron Lett. 1991, 32, 6371–6374. doi:10.1016/0040-4039(91)80172-3
Return to citation in text: [1] -
Matsuda, F.; Sakai, T.; Okada, N.; Miyashita, M. Tetrahedron Lett. 1998, 39, 863–864. doi:10.1016/S0040-4039(97)10750-X
Return to citation in text: [1] -
Tamiya, H.; Goto, K.; Matsuda, F. Org. Lett. 2004, 6, 545–547. doi:10.1021/ol036329h
Return to citation in text: [1] -
Khan, F. A.; Czerwonka, R.; Zimmer, R.; Reissig, H.-U. Synlett 1997, 995–997. doi:10.1055/s-1997-934
Return to citation in text: [1] [2] [3] -
Nandanan, E.; Dinesh, C. U.; Reissig, H.-U. Tetrahedron 2000, 56, 4267–4277. doi:10.1016/S0040-4020(00)00353-7
Return to citation in text: [1] [2] -
Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Saadi, J.; Reissig, H.-U. Synlett 2009, 2089–2092. doi:10.1055/s-0029-1217520
Return to citation in text: [1] [2] -
Saadi, J.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 3334–3337. doi:10.1021/ol901183h
Return to citation in text: [1] [2] -
Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 512–530. doi:10.1002/jlac.198419840311
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n
Return to citation in text: [1] -
Suzuki, A. J. Organomet. Chem. 1999, 576, 147–168. doi:10.1016/S0022-328X(98)01055-9
Return to citation in text: [1] -
Molander, G. A.; Rivero, M. R. Org. Lett. 2002, 4, 107–109. doi:10.1021/ol0169729
Return to citation in text: [1] -
Curran, D. P.; Wolin, R. L. Synlett 1991, 317–318. doi:10.1055/s-1991-34728
Return to citation in text: [1] -
Miller, R. S.; Sealy, J. M.; Shabangi, M.; Kuhlman, M. L.; Fuchs, J. R.; Flowers, R. A., II. J. Am. Chem. Soc. 2000, 122, 7718–7722. doi:10.1021/ja001260j
Return to citation in text: [1] -
Inokuchi, T. J. Org. Chem. 2005, 70, 1497–1500. doi:10.1021/jo0402529
Return to citation in text: [1] -
Sadasivam, D. V.; Antharjanam, P. K. S.; Prasad, E.; Flowers, R. A., II. J. Am. Chem. Soc. 2008, 130, 7228–7229. doi:10.1021/ja802448x
Return to citation in text: [1] -
Flowers, R. A., II. Synlett 2008, 1427–1439. doi:10.1055/s-2008-1078414
Return to citation in text: [1] -
Seebach, D.; Prelog, V. Angew. Chem., Int. Ed. Engl. 1982, 21, 654–660. doi:10.1002/anie.198206541
Return to citation in text: [1] -
Parmar, D.; Duffy, L. A.; Sadasivam, D. V.; Matsubara, H.; Bradley, P. A.; Flowers, R. A., II; Procter, D. J. J. Am. Chem. Soc. 2009, 131, 15467–15473. doi:10.1021/ja906396u
Return to citation in text: [1] -
Haque, A.; Ghosh, S. J. Chem. Soc., Chem. Commun. 1997, 2039–2040. doi:10.1039/a705843h
Return to citation in text: [1] -
Williams, D. B. G.; Blann, K.; Holzapfel, C. W. J. Org. Chem. 2000, 65, 2834–2836. doi:10.1021/jo9919005
Return to citation in text: [1] -
Schwartz, A.; Seger, C. Monatsh. Chem. 2001, 132, 855–858. doi:10.1007/s007060170074
Return to citation in text: [1] -
Williams, D. B. G.; Blann, K.; Holzapfel, C. W. J. Chem. Soc., Perkin Trans. 1 2001, 219–220. doi:10.1039/b008110h
Return to citation in text: [1] -
CCDC-789452 (for 17b) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.ac.uk/data_request/cif.
Return to citation in text: [1] -
Diamond software, 2.1d; CRYSTAL IMPACT, GbR: Bonn, Germany.
Return to citation in text: [1] [2] -
Dinesh, C. U.; Reissig, H.-U. Angew. Chem., Int. Ed. 1999, 38, 789–791. doi:10.1002/(SICI)1521-3773(19990315)38:6<789::AID-ANIE789>3.0.CO;2-W
Return to citation in text: [1] -
Berndt, M.; Reissig, H.-U. Synlett 2001, 1290–1292. doi:10.1055/s-2001-16051
Return to citation in text: [1] -
Gross, S.; Reissig, H.-U. Synlett 2002, 2027–2030. doi:10.1055/s-2002-35599
Return to citation in text: [1] -
Aulenta, F.; Berndt, M.; Brüdgam, I.; Hartl, H.; Sörgel, S.; Reissig, H.-U. Chem.–Eur. J. 2007, 13, 6047–6062. doi:10.1002/chem.200700057
Return to citation in text: [1] -
Wefelscheid, U. K.; Berndt, M.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3635–3646. doi:10.1002/ejoc.200800293
Return to citation in text: [1] [2] [3] -
Beemelmanns, C.; Blot, V.; Gross, S.; Lentz, D.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2716–2732. doi:10.1002/ejoc.200901455
Return to citation in text: [1] -
CCDC-789453 (for 40) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.ac.uk/data_request/cif.
Return to citation in text: [1] -
Namy, J.-L.; Kagan, H. B. Nouv. J. Chim. 1977, 1, 5–7.
Return to citation in text: [1] -
Girard, P.; Namy, J.-L.; Kagan, H. B. J. Am. Chem. Soc. 1980, 102, 2693–2698. doi:10.1021/ja00528a029
Return to citation in text: [1] -
Imamoto, T.; Ono, M. Chem. Lett. 1987, 501–502. doi:10.1246/cl.1987.501
Return to citation in text: [1] -
Molander, G. A.; del Pozo Losada, C. Tetrahedron 1998, 54, 5819–5832. doi:10.1016/S0040-4020(98)00272-5
Return to citation in text: [1]
| 51. | CCDC-789452 (for 17b) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.ac.uk/data_request/cif. |
| 20. | Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429 |
| 53. | Dinesh, C. U.; Reissig, H.-U. Angew. Chem., Int. Ed. 1999, 38, 789–791. doi:10.1002/(SICI)1521-3773(19990315)38:6<789::AID-ANIE789>3.0.CO;2-W |
| 54. | Berndt, M.; Reissig, H.-U. Synlett 2001, 1290–1292. doi:10.1055/s-2001-16051 |
| 55. | Gross, S.; Reissig, H.-U. Synlett 2002, 2027–2030. doi:10.1055/s-2002-35599 |
| 56. | Aulenta, F.; Berndt, M.; Brüdgam, I.; Hartl, H.; Sörgel, S.; Reissig, H.-U. Chem.–Eur. J. 2007, 13, 6047–6062. doi:10.1002/chem.200700057 |
| 57. | Wefelscheid, U. K.; Berndt, M.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3635–3646. doi:10.1002/ejoc.200800293 |
| 58. | Beemelmanns, C.; Blot, V.; Gross, S.; Lentz, D.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2716–2732. doi:10.1002/ejoc.200901455 |
| 1. | Mehta, G.; Singh, V. Chem. Rev. 1999, 99, 881–930. doi:10.1021/cr9800356 |
| 2. | Petasis, N. A.; Patane, M. A. Tetrahedron 1992, 48, 5757–5821. doi:10.1016/S0040-4020(01)90172-3 |
| 5. | Fujiwara, K.; Goto, A.; Sato, D.; Kawai, H.; Suzuki, T. Tetrahedron Lett. 2005, 46, 3465–3468. doi:10.1016/j.tetlet.2005.03.114 |
| 31. | Khan, F. A.; Czerwonka, R.; Zimmer, R.; Reissig, H.-U. Synlett 1997, 995–997. doi:10.1055/s-1997-934 |
| 4. | Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. doi:10.1002/anie.200500369 |
| 35. | Saadi, J.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 3334–3337. doi:10.1021/ol901183h |
| 1. | Mehta, G.; Singh, V. Chem. Rev. 1999, 99, 881–930. doi:10.1021/cr9800356 |
| 2. | Petasis, N. A.; Patane, M. A. Tetrahedron 1992, 48, 5757–5821. doi:10.1016/S0040-4020(01)90172-3 |
| 4. | Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. doi:10.1002/anie.200500369 |
| 5. | Fujiwara, K.; Goto, A.; Sato, D.; Kawai, H.; Suzuki, T. Tetrahedron Lett. 2005, 46, 3465–3468. doi:10.1016/j.tetlet.2005.03.114 |
| 6. | Nair, V.; Abhilash, K. G. Synlett 2008, 301–312. doi:10.1055/s-2008-1032058 |
| 7. | Ohno, H.; Hamaguchi, H.; Ohata, M.; Kosaka, S.; Tanaka, T. J. Am. Chem. Soc. 2004, 126, 8744–8754. doi:10.1021/ja048693x |
| 8. | Yu, Z.-X.; Wang, Y.; Wang, Y. Chem.–Asian J. 2010, 5, 1072–1088. doi:10.1002/asia.200900712 |
| 9. | Genrich, F.; Schaumann, E. Tetrahedron Lett. 2009, 50, 6187–6190. doi:10.1016/j.tetlet.2009.08.092 |
| 10. | Zhang, T.; Huang, X.; Xue, J.; Sun, S. Tetrahedron Lett. 2009, 50, 1290–1294. doi:10.1016/j.tetlet.2009.01.001 |
| 11. | Evans, P. A.; Holmes, B. Tetrahedron 1991, 47, 9131–9166. doi:10.1016/S0040-4020(01)96203-9 |
| 12. | Roxburgh, C. J. Tetrahedron 1993, 49, 10749–10784. doi:10.1016/S0040-4020(01)80232-5 |
| 13. | Rousseau, G. Tetrahedron 1995, 51, 2777–2849. doi:10.1016/0040-4020(94)01064-7 |
| 14. | Hoberg, J. O. Tetrahedron 1998, 54, 12631–12670. doi:10.1016/S0040-4020(98)00596-1 |
| 15. | Yet, L. Tetrahedron 1999, 55, 9349–9403. doi:10.1016/S0040-4020(99)00483-4 |
| 16. | Yet, L. Chem. Rev. 2000, 100, 2963–3008. doi:10.1021/cr990407q |
| 17. | Nubbemeyer, U. Top. Curr. Chem. 2001, 216, 125–196. doi:10.1007/3-540-44726-1_4 |
| 18. | Shiina, I. Chem. Rev. 2007, 107, 239–273. doi:10.1021/cr050045o |
| 20. | Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429 |
| 32. | Nandanan, E.; Dinesh, C. U.; Reissig, H.-U. Tetrahedron 2000, 56, 4267–4277. doi:10.1016/S0040-4020(00)00353-7 |
| 60. | Namy, J.-L.; Kagan, H. B. Nouv. J. Chim. 1977, 1, 5–7. |
| 61. | Girard, P.; Namy, J.-L.; Kagan, H. B. J. Am. Chem. Soc. 1980, 102, 2693–2698. doi:10.1021/ja00528a029 |
| 3. | Galli, C.; Mandolini, L. Eur. J. Org. Chem. 2000, 3117–3125. doi:10.1002/1099-0690(200009)2000:18<3117::AID-EJOC3117>3.0.CO;2-5 |
| 34. | Saadi, J.; Reissig, H.-U. Synlett 2009, 2089–2092. doi:10.1055/s-0029-1217520 |
| 62. | Imamoto, T.; Ono, M. Chem. Lett. 1987, 501–502. doi:10.1246/cl.1987.501 |
| 63. | Molander, G. A.; del Pozo Losada, C. Tetrahedron 1998, 54, 5819–5832. doi:10.1016/S0040-4020(98)00272-5 |
| 10. | Zhang, T.; Huang, X.; Xue, J.; Sun, S. Tetrahedron Lett. 2009, 50, 1290–1294. doi:10.1016/j.tetlet.2009.01.001 |
| 22. | Molander, G. A.; McKie, J. A. J. Org. Chem. 1994, 59, 3186–3192. doi:10.1021/jo00090a041 |
| 23. | Molander, G. A.; Alonso-Alija, C. J. Org. Chem. 1998, 63, 4366–4373. doi:10.1021/jo980119e |
| 24. | Molander, G. A.; Machrouhi, F. J. Org. Chem. 1999, 64, 4119–4123. doi:10.1021/jo990216n |
| 25. | Molander, G. A.; Köllner, C. J. Org. Chem. 2000, 65, 8333–8339. doi:10.1021/jo001195w |
| 26. | Molander, G. A.; Le Huérou, Y.; Brown, G. A. J. Org. Chem. 2001, 66, 4511–4516. doi:10.1021/jo001513r |
| 27. | Molander, G. A.; Brown, G. A.; Storch de Gracia, I. J. Org. Chem. 2002, 67, 3459–3463. doi:10.1021/jo020027w |
| 28. | Inanaga, J.; Yokoyama, Y.; Handa, Y.; Yamaguchi, M. Tetrahedron Lett. 1991, 32, 6371–6374. doi:10.1016/0040-4039(91)80172-3 |
| 29. | Matsuda, F.; Sakai, T.; Okada, N.; Miyashita, M. Tetrahedron Lett. 1998, 39, 863–864. doi:10.1016/S0040-4039(97)10750-X |
| 30. | Tamiya, H.; Goto, K.; Matsuda, F. Org. Lett. 2004, 6, 545–547. doi:10.1021/ol036329h |
| 31. | Khan, F. A.; Czerwonka, R.; Zimmer, R.; Reissig, H.-U. Synlett 1997, 995–997. doi:10.1055/s-1997-934 |
| 32. | Nandanan, E.; Dinesh, C. U.; Reissig, H.-U. Tetrahedron 2000, 56, 4267–4277. doi:10.1016/S0040-4020(00)00353-7 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 34. | Saadi, J.; Reissig, H.-U. Synlett 2009, 2089–2092. doi:10.1055/s-0029-1217520 |
| 35. | Saadi, J.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 3334–3337. doi:10.1021/ol901183h |
| 9. | Genrich, F.; Schaumann, E. Tetrahedron Lett. 2009, 50, 6187–6190. doi:10.1016/j.tetlet.2009.08.092 |
| 20. | Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429 |
| 31. | Khan, F. A.; Czerwonka, R.; Zimmer, R.; Reissig, H.-U. Synlett 1997, 995–997. doi:10.1055/s-1997-934 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 57. | Wefelscheid, U. K.; Berndt, M.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3635–3646. doi:10.1002/ejoc.200800293 |
| 7. | Ohno, H.; Hamaguchi, H.; Ohata, M.; Kosaka, S.; Tanaka, T. J. Am. Chem. Soc. 2004, 126, 8744–8754. doi:10.1021/ja048693x |
| 8. | Yu, Z.-X.; Wang, Y.; Wang, Y. Chem.–Asian J. 2010, 5, 1072–1088. doi:10.1002/asia.200900712 |
| 57. | Wefelscheid, U. K.; Berndt, M.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3635–3646. doi:10.1002/ejoc.200800293 |
| 19. | Molander, G. A. Acc. Chem. Res. 1998, 31, 603–609. doi:10.1021/ar960101v |
| 20. | Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429 |
| 21. | Procter, D. J.; Flowers, R. A., II; Skrydstrup, T. Organic Synthesis using Samarium Diiodide; Royal Society of Chemistry: Cambridge, U.K., 2009. |
| 59. | CCDC-789453 (for 40) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.ac.uk/data_request/cif. |
| 38. | Suzuki, A. J. Organomet. Chem. 1999, 576, 147–168. doi:10.1016/S0022-328X(98)01055-9 |
| 39. | Molander, G. A.; Rivero, M. R. Org. Lett. 2002, 4, 107–109. doi:10.1021/ol0169729 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 36. | Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 512–530. doi:10.1002/jlac.198419840311 |
| 37. | Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n |
| 46. | Parmar, D.; Duffy, L. A.; Sadasivam, D. V.; Matsubara, H.; Bradley, P. A.; Flowers, R. A., II; Procter, D. J. J. Am. Chem. Soc. 2009, 131, 15467–15473. doi:10.1021/ja906396u |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 47. | Haque, A.; Ghosh, S. J. Chem. Soc., Chem. Commun. 1997, 2039–2040. doi:10.1039/a705843h |
| 48. | Williams, D. B. G.; Blann, K.; Holzapfel, C. W. J. Org. Chem. 2000, 65, 2834–2836. doi:10.1021/jo9919005 |
| 49. | Schwartz, A.; Seger, C. Monatsh. Chem. 2001, 132, 855–858. doi:10.1007/s007060170074 |
| 50. | Williams, D. B. G.; Blann, K.; Holzapfel, C. W. J. Chem. Soc., Perkin Trans. 1 2001, 219–220. doi:10.1039/b008110h |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 45. | Seebach, D.; Prelog, V. Angew. Chem., Int. Ed. Engl. 1982, 21, 654–660. doi:10.1002/anie.198206541 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
| 40. | Curran, D. P.; Wolin, R. L. Synlett 1991, 317–318. doi:10.1055/s-1991-34728 |
| 41. | Miller, R. S.; Sealy, J. M.; Shabangi, M.; Kuhlman, M. L.; Fuchs, J. R.; Flowers, R. A., II. J. Am. Chem. Soc. 2000, 122, 7718–7722. doi:10.1021/ja001260j |
| 42. | Inokuchi, T. J. Org. Chem. 2005, 70, 1497–1500. doi:10.1021/jo0402529 |
| 43. | Sadasivam, D. V.; Antharjanam, P. K. S.; Prasad, E.; Flowers, R. A., II. J. Am. Chem. Soc. 2008, 130, 7228–7229. doi:10.1021/ja802448x |
| 44. | Flowers, R. A., II. Synlett 2008, 1427–1439. doi:10.1055/s-2008-1078414 |
| 33. | Reissig, H.-U.; Khan, F. A.; Czerwonka, R.; Dinesh, C. U.; Shaikh, A. L.; Zimmer, R. Eur. J. Org. Chem. 2006, 4419–4428. doi:10.1002/ejoc.200600360 |
© 2010 Saadi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)