
Abstract
A series of polycyclic frameworks with fluorinated syn-facial quinoxaline sidewalls has been prepared as potential molecular tweezers for electron-rich guest compounds. Our synthetic route to the cyclooctadiene-derived scaffolds 16a–d takes advantage of the facile isolation of a novel spirocyclic precursor 9b with the crucial syn-orientation of its two alkene moieties. The crystal structure of 16c displays two features typical of a molecular tweezer: inclusion of a solvent molecule in the molecular cleft and self-association of the self-complementary scaffolds. Furthermore, host–guest NMR studies of compound 16c in solution show chemical exchange between the unbound and bound electron-rich guest, N,N,N′,N′-tetramethyl-p-phenylenediamine.
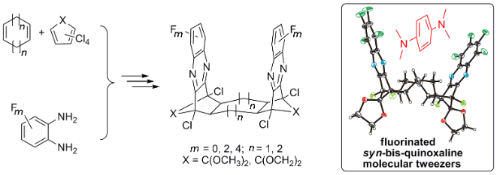
Graphical Abstract
Introduction
A broad variety of structurally diverse molecular tweezers, i.e., scaffolds in which a tether unit connects two syn-oriented aromatic pincers, are well-established as devices for the molecular recognition of mostly electron-deficient guest compounds [1-10]. Conversely, molecular tweezers with a binding cleft that displays an inverted electrostatic potential could thus find application in sensing of electron-rich guests, or even anions [11-13]. Possible frameworks include the seemingly trivial fluorinated analogues of known frameworks (Scheme 1), but so far only a few groups have investigated these intriguing target compounds: Korenaga and Sakai optimized the synthetic access to fluorinated acridine-based molecular tweezers 1 and determined association constants for the complexation of electron-rich arenes [14,15]. Hermida-Ramón and Estévez calculated the structures and electrostatic potentials of belt-shaped compounds 2a–c and predicted the complexation of halide anions in the cavity of 2c [16-18].
![[1860-5397-6-39-i1]](/bjoc/content/inline/1860-5397-6-39-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fluorinated molecular tweezers.
Scheme 1: Fluorinated molecular tweezers.
Intrigued by Chou’s communication on the spectroscopic properties of non-fluorinated bis-quinoxalines of type 3 and 4a [19], we targeted on the corresponding fluorinated derivatives – in particular compound 4b with its large binding cleft.
In this paper, we present the synthesis and characterization of these synthetically more challenging derivatives. Furthermore, we discuss structural features of a cyclooctadiene-derived scaffold of type 4b and report preliminary spectroscopic data on their association with electron-rich guest compounds.
Results and Discussion
Synthesis of fluorinated bis-quinoxalines
The general route [19] to bis-quinoxaline targets (Scheme 2) utilizes a twofold Diels–Alder reaction of a cycloalkadiene (5,6) with cyclopentadienone derivatives (7), subsequent oxidation of the syn-diene intermediates (8,9) to their corresponding tetraketones (10,11) and condensation of the latter with o-phenylenediamine derivatives (12) to obtain the syn-bis-quinoxaline target compounds (15,16). This synthetic route is flexible with regard to the tether size (cyclohexane vs cyclooctane) and modifications in the pincer sidewalls (degree of fluorination).
![[1860-5397-6-39-i2]](/bjoc/content/inline/1860-5397-6-39-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of non-fluorinated and fluorinated syn-bis-quinoxalines.
Scheme 2: Synthesis of non-fluorinated and fluorinated syn-bis-quinoxalines.
Although only the larger cyclooctadiene-derived scaffolds 16a–d could function as molecular tweezers, we also synthesized the fluorinated cyclohexadiene-derived compounds 15b–d with their smaller π-π–distances. A Diels–Alder reaction of cyclohexadiene (5) with ketal 7a furnished exclusively the syn-bis-adduct 8a [20] which was then converted to the canary-yellow tetraketone 10 by Khan’s original RuCl3-catalyzed oxidation protocol [21-23] since Chou’s “optimized” procedure was somewhat capricious in our hands. The twofold condensation with di- or tetrafluoro-o-phenylenediamine (12b,c) [24,25] provided access to the novel fluorinated species 15b–c in acceptable yields (60–70%). This last reaction required harsh conditions and delivered a dark crude product with unspecified tarry material after heating the substrates for several days to 115 °C (1H and 19F NMR control). Occasionally, the condensation reaction did not lead to complete conversion of the tetraketone precursor 10 and produced a separable mixture of the mono- and bis-condensation products 13 and 15, respectively. The isolated mono-adducts 13a (or 13b) could then be converted to the symmetrical target 15a (or 15c) or, upon condensation with the appropriate o-phenylenediamine derivative 12c (or 12a), to scaffold 15b with only one fluorinated quinoxaline subunit.
The synthetic access to cyclooctadiene-derived scaffolds is complicated by the lack of selectivity in the twofold Diels–Alder reaction of diene 6 and led to a mixture of the syn- and anti-bis-adducts in a 1:4 ratio [26,27]. Since the separation of the crucial syn-isomer 9a from anti-compound 9a′ by repeated recrystallization did not furnish the pure endo,endo,syn-isomer 9a in our hands, we focused on the new spirocyclic derivative 9b. Thus, reaction of the spiro-ketal 7b [28] with cyclooctadiene (6) furnished a mixture of 9b and 9b′ in excellent yield in the same ratio of isomers as observed in the previous case. Again, the endo,endo,syn-isomer 9b could not be satisfactorily separated from the endo,endo,anti-isomer 9b′ by chromatography, but gram-amounts of the crucial syn-isomer 9b were readily obtained after repeated recrystallization from hot diethyl ether. The assignment of both syn- and anti-isomers was initially based on 1H NMR spectroscopic analogies to the bis-methoxyketals, i.e., the small low-frequency shift of the bridgehead proton resonances of the anti-adduct (Δδ = 0.20 ppm). The X-ray structure determination of target compound 16c confirmed indirectly the correct assignment of isomers 9b and 9b′ (vide infra). Oxidation of 9b with Khan’s original protocol [21-23] and condensation of the resulting tetraketone 11 with o-phenylenediamine 12a or the fluorinated derivatives 12b–c resulted in the new non-fluorinated parent compound 16a and the three fluorinated scaffolds 16b–c, respectively. All new syn-bis-quinoxalines were purified by flash-chromatography on silica gel and obtained as off-white powders in 60–75% yield after recrystallization from methanol. Considering the low nucleophilicity of the fluorinated amine building blocks 12b–c, our yields in the condensation reaction are quite good (71–86% for each condensation step) and any modification of the reaction conditions by other reported procedures [29-32] did not significantly alter the outcome. It should be noted that all fluorinated bis-quinoxalines are stable compounds which do not show any decomposition over extended periods of time; loss of fluorine has only been observed under typical nucleophilic aromatic substitution conditions.
Although the new compounds, in particular the cyclohexadiene-derived species 15b–c, were reasonably soluble in dipolar aprotic solvents (DMSO, DMF) or halogenated aromatic solvents (C6H5Cl), they only displayed poor solubility in several standard organic solvents (CHCl3, CH2Cl2, CH3OH, C6H6, CH3CN). Their full characterization and some preliminary host–guest studies of the cyclooctadiene-derived frameworks could however, be carried out in dilute chloroform, acetonitrile and methylene chloride solutions. The spectroscopic characteristics of 15a and several non-fluorinated derivatives have been described elsewhere [19] and the NMR spectroscopic data for 15b–c (16a–d) are only altered by the absence of the corresponding proton resonances, the additional coupling of fluorine with either the arene protons in 15c (16c) or the aromatic carbon atoms, and the more complex signal structure of the spirocyclic ketal in 16a–d. The UV–vis spectra (available in the Supporting Information File 1) display the expected electronic transitions for quinoxaline derivatives [33-35], i.e., a prominent π,π* transition with λmax between 236–245 nm and a lower intensity n,π* transition with λmax between 312–316 nm with a poorly resolved vibrational structure. The spectra of the cyclohexadiene-derived scaffolds 15 and the cyclooctadiene-derived frameworks 16 are very similar. Within each series we could not observe a gradual blue-shift for the electronic transitions as the degree of fluorination increased from 15a (16a) to 15c (16c), a result that is in accord with Chou’s UV–vis data for differently substituted bis-quinoxaline scaffolds that abstain from clear trends as the electronic-withdrawing character of the substituents were altered [19]. The ESI-mass spectra (acetonitrile, acetic acid) of all new syn-bis-quinoxalines show the correct isotopic pattern of the protonated molecules and, interestingly, display mass clusters for the protonated “dimers” of compounds 15b and 16b. Nevertheless, any interpretation of the nature of these latter species (proton-bridged “dimer”, protonated π-π–aggregate, protonated self-associated “dimer”) requires further investigation and cannot be easily transferred to the solution- or solid-state structures of the neutral tweezer compounds [36].
Structures
We were able to grow single crystals of the octafluoro compound 16c from acetonitrile or chloroform solutions suitable for X-ray structure determination (Table 1, Figure 1). In each case, the crystals contained residual ethyl acetate from the purification step, indicating strong binding of the ethyl acetate molecule inside the binding cleft of 16c. Compound 16c crystallizes, with an ethyl acetate solvent molecule, in the monoclinic system (space group: P21/n) and displays bond lengths and angles in the expected ranges. The ethyl acetate displays a small degree of orientation disorder (11.8%). Figure 1a shows a thermal ellipsoid image of 16c and Figure 1b depicts the packing within a unit cell setting. The large binding pocket of syn-bis-quinoxaline 16c provides enough space to allow the association with solvent (ethyl acetate) and, through “dimer formation”, with the pincer sidewall of a second tweezer molecule. The “dimer” association of fluorinated molecular tweezers in the solid state has been observed for the acridine-derived scaffold 1 [15] and is quite common in many other molecular tweezer scaffolds [1-10]. Compound 16c shows the typical orientation of fluorine substituents of one pincer sidewall over the arene subunit of another tweezer (substituent distances to arene plane: 3.283 Å, 3.315 Å), interpreted as the attractive interaction between fluorine substituents with the electron-depleted fluoroarene subunit [37,38]. The centroid-centroid distances (d1, d2) and the bite angle between the two quinoxaline sidewalls of the binding pocket in fluorinated framework 16c differ only slightly from the parameters of the non-fluorinated compound 4a, although the latter does not include any solvent in the cleft and, furthermore, lacks the interpenetrating self-association displayed in 16c [19]. Conversely, 4a shows π-π–interaction of two adjacent molecules by stacking two pincer sidewalls, each from the outside (U···U geometry).
![[1860-5397-6-39-1]](/bjoc/content/figures/1860-5397-6-39-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Thermal ellipsoid image of the tweezer molecular 16c in the structure 16c · CH3CO2C2H5; thermal ellipsoids are drawn at the 50% probability level. (b) View of the packing of 16c in the unit cell (two CH3CO2C2H5 molecules are omitted for clarity). [C34H20Cl4F8N4O4 · CH3CO2C2H5, MW = 930.44, monoclinic, space group P21/n, a = 15.3990(12) Å, b = 14.0635(11) Å, c = 17.7148(14) Å, β = 94.6470(10)°, V = 3823.8(5) Å3, Z = 4, dcalc = 1.616 g cm−3, T = 153(2) K, λ = 0.71073 Å, 44247 reflections measured, 9378 unique (Rint = 0.017), final R1 [I > 2δ(I)] = 0.0343 and R1 = 0.0403 (wR2 = 0.0952) for all data; CCDC deposit # 786086.
Figure 1: (a) Thermal ellipsoid image of the tweezer molecular 16c in the structure 16c · CH3CO2C2H5; thermal...
Table 1: Crystallographic details for 16c and related non-fluorinated compounds.
adefined in Scheme 2.
ba negative bite angle defines U- vs. V-shaped tweezers.
Host–Guest Chemistry
Although none of the reported cyclooctadiene-derived syn-bis-quinoxaline scaffolds [19] has been established as a molecular tweezer, the general architecture with two syn-oriented aromatic sidewalls and a large π-π–distance does allow the accommodation of guest compounds as demonstrated in the crystal structure of 16c. Whilst most molecular tweezers have a typical cleft size of ca. 7 Å, several functional larger systems have been reported [39,40]. Figure 2 shows the electrostatic potential surfaces of compounds 16a–c, depicting the inversion of the electrostatic potential in the pincer subunits upon increasing the degree of fluorination.
![[1860-5397-6-39-2]](/bjoc/content/figures/1860-5397-6-39-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Electrostatic potential surfaces of 16a–c (Spartan 06 [41]: B3LYP/6-31G*//B3LYP/6-31G*; legend in kcal/mol).
Figure 2: Electrostatic potential surfaces of 16a–c (Spartan 06 [41]: B3LYP/6-31G*//B3LYP/6-31G*; legend in kcal/...
NMR titration experiments with electron-rich arenes (1,4-dimethoxybenzene, 1,3,5-trimethoxybenzene, N,N-dimethylaniline, N,N,N′,N′-tetramethyl-p-phenylenediamine) were carried out in deuterated methylene chloride solution for the four cyclooctadiene-derived species 16a–d. Interestingly, only the octafluoro-derivative 16c showed line-broadening of the 1H resonances for one guest compound, i.e., N,N,N′,N′-tetramethyl-p-phenylenediamine, at various host–guest ratios (Figure 3). No changes in chemical shift of the quinoxaline 19F resonances were observed in the 19F NMR spectra. Upon cooling the NMR samples the guest’s aromatic and methyl 1H resonances sharpened only to less broad signals. Titration of 16c with other electron-rich aromatic guest compounds (1,4-dimethoxybenzene, 1,3,5-trimethoxybenzene, N,N-dimethylaniline) under the same conditions showed only the original host and guest resonances in the 1H NMR spectra without any line broadening, which indicates that there was no interaction between these three molecules with the tweezer’s cavity. It is important to note that from the entire series of compounds, only the highly fluorinated scaffold 16c shows chemical exchange between the unbound and bound guest, N,N,N′,N′-tetramethyl-p-phenylenediamine. While this facile exchange is certainly due to the large binding cleft, the effect of eight fluorine substituents on the electrostatic potential within the cleft is paramount in the facilitation of this interaction between host and guest.
![[1860-5397-6-39-3]](/bjoc/content/figures/1860-5397-6-39-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra (CD2Cl2, 500 MHz) of 16c (host [black]) upon titration with N,N,N′,N′-tetramethyl-p-phenylenediamine (guest [red]); host concentration: 0.01M; host–guest ratio: 1:2 (1), 1:5 (2), 1:10 (3), 1:20 (4).
Figure 3: 1H NMR spectra (CD2Cl2, 500 MHz) of 16c (host [black]) upon titration with N,N,N′,N′-tetramethyl-p-...
Korenaga and Sakai have already noted that N,N,N′,N′-tetramethyl-p-phenylendiamine displays a stronger association constant with molecular tweezer 1 when compared to several other electron-rich aromatic guest compounds. This behavior was explained by the large magnitude of the former guest’s quadrupole moment [15].
With our preliminary NMR titrations we could demonstrate that scaffold 16 can indeed associate with an external guest compound in solution if the host and guest units are matched appropriately. Further experiments employing complementary analytical techniques, e.g., isothermal calorimetry, as well as additional investigations of the host–guest chemistry with suitable, larger guest compounds, will provide detailed thermodynamic parameters of the host–guest association, and possibly a better host–guest match, respectively.
Conclusion
The synthesis of fluorinated syn-bis-quinoxalines (15b–c, 16b–c) was successfully accomplished by a three-step procedure, utilizing the new, readily isolable spirocyclic syn-derivative 9b as an entry towards the larger cyclooctadiene-derived scaffold 16. The crystal structure of 16c clearly demonstrates that syn-bis-quinoxaline frameworks can function as molecular tweezers. Furthermore, preliminary NMR spectroscopic titration experiments with the octafluoro-syn-bis-quinoxaline 16c prove the interaction of an external, electron-rich guest with the molecular tweezer’s cavity in solution.
References
-
Chen, C.-W.; Whitlock, H. W., Jr. J. Am. Chem. Soc. 1978, 100, 4921–4922. doi:10.1021/ja00483a063
Return to citation in text: [1] [2] -
Zimmerman, S. C. Top. Curr. Chem. 1993, 165, 71–102. doi:10.1007/BFb0111281
Return to citation in text: [1] [2] -
Klärner, F.-G.; Kahlert, B. Acc. Chem. Res. 2003, 36, 919–932. doi:10.1021/ar0200448
Return to citation in text: [1] [2] -
Harmata, M. Acc. Chem. Res. 2004, 37, 862–873. doi:10.1021/ar030164v
Return to citation in text: [1] [2] -
Rowan, A. E.; Elemans, J. A. A. W.; Nolte, R. J. M. Acc. Chem. Res. 1999, 32, 995–1006. doi:10.1021/ar9702684
Return to citation in text: [1] [2] -
Katz, J. L.; Geller, B. J.; Foser, P. D. Chem. Commun. 2007, 1026–1028. doi:10.1039/b615336d
Return to citation in text: [1] [2] -
Wei, T.-B.; Wei, W.; Cao, C.; Zhang, Y.-M. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 1218–1228. doi:10.1080/10426500701613154
Return to citation in text: [1] [2] -
Bhattacharya, S.; Tominaga, K.; Kimura, T.; Uno, H.; Komatsu, N. Chem. Phys. Lett. 2007, 433, 395–402. doi:10.1016/j.cplett.2006.11.039
Return to citation in text: [1] [2] -
Skibinski, M.; Gómez, R.; Lork, E.; Azov, V. A. Tetrahedron 2009, 65, 10348–10354. doi:10.1016/j.tet.2009.10.052
Return to citation in text: [1] [2] -
Nishiuchi, T.; Kuwatani, Y.; Nishinaga, T.; Iyoda, M. Chem.–Eur. J. 2009, 15, 6838–6847. doi:10.1002/chem.200900623
Return to citation in text: [1] [2] -
Hay, B. P.; Custelcean, R. Cryst. Growth Des. 2009, 9, 2593–2545. doi:10.1021/cg900308b
Return to citation in text: [1] -
Schottel, B. L.; Chifotides, H. T.; Dunbar, K. R. Chem. Soc. Rev. 2008, 37, 68–83. doi:10.1039/b614208g
Return to citation in text: [1] -
Fujita, M.; Nagao, S.; Iida, M.; Ogata, K.; Ogura, K. J. Am. Chem. Soc. 1993, 115, 1574–1576. doi:10.1021/ja00057a052
Return to citation in text: [1] -
Korenaga, T.; Kosaki, T.; Kawauchi, Y.; Ema, T.; Sakai, T. J. Fluorine Chem. 2006, 127, 604–609. doi:10.1016/j.jfluchem.2005.12.007
Return to citation in text: [1] -
Korenaga, T.; Kawauchi, Y.; Kosaki, T.; Ema, T.; Sakai, T. Bull. Chem. Soc. Jpn. 2005, 78, 2175–2179. doi:10.1246/bcsj.78.2175
Return to citation in text: [1] [2] [3] -
Mandado, M.; Hermida-Ramón, J. M.; Estévez, C. M. THEOCHEM 2006, 854, 1–9.
Return to citation in text: [1] -
Hermida-Ramón, J. M.; Estévez, C. M. Chem.–Eur. J. 2007, 13, 4743–4749. doi:10.1002/chem.200601836
Return to citation in text: [1] -
Hermida-Ramón, J. M.; Mandado, M.; Sánchez-Lozano, M.; Estévez, C. M. Phys. Chem. Chem. Phys. 2010, 12, 164–169. doi:10.1039/b915483c
Return to citation in text: [1] -
Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Chou, T.-C.; Yang, M.-S.; Lin, C.-T. J. Org. Chem. 1994, 59, 661–663. doi:10.1021/jo00082a028
Return to citation in text: [1] -
Khan, F. A.; Dash, J.; Sudheer, C.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385
Return to citation in text: [1] [2] -
Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424–2425. doi:10.1021/ja017371f
Return to citation in text: [1] [2] -
Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558–9559. doi:10.1021/ja001956c
Return to citation in text: [1] [2] -
Arotsky, J.; Butler, R.; Darby, A. C. J. Chem. Soc. C 1970, 1480–1485. doi:10.1039/J39700001480
Return to citation in text: [1] -
Heaton, M.; Hill, P.; Drakesmith, F. J. Fluorine Chem. 1997, 81, 133–138. doi:10.1016/S0022-1139(96)03517-8
Return to citation in text: [1] -
Garcia, J. G.; Fronczek, F. R.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3289–3292. doi:10.1016/S0040-4039(00)92688-1
Return to citation in text: [1] -
Garcia, J. G.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3293–3296. doi:10.1016/S0040-4039(00)92689-3
Return to citation in text: [1] -
Mackenzie, K. J. Chem. Soc. 1964, 5710–5716. doi:10.1039/JR9640005710
Return to citation in text: [1] -
Azizian, J.; Karimi, A. R.; Kazemizadeh, Z.; Mohammadi, A. A.; Mohammadizadeh, M. R. J. Org. Chem. 2005, 70, 1471–1473. doi:10.1021/jo0486692
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Rao, Y. G.; Narasaiah, A. V. Chem. Lett. 2008, 37, 348–349. doi:10.1246/cl.2008.348
Return to citation in text: [1] -
Sangshetti, J. N.; Kokare, N. D.; Shinde, D. B. Russ. J. Org. Chem. 2009, 45, 1116–1118. doi:10.1134/S1070428009070240
Return to citation in text: [1] -
Zhao, Z.; Wisnoski, D.; Wolkenberg, S. E.; Leister, W. H.; Wang, Y.; Lindsley, C. W. Tetrahedron Lett. 2004, 45, 4873–4876. doi:10.1016/j.tetlet.2004.04.144
Return to citation in text: [1] -
Waluk, J.; Grabowska, A.; Pakulstroka, B. J. Lumin. 1980, 21, 277–291. doi:10.1016/0022-2313(80)90007-1
Return to citation in text: [1] -
Imes, K. K.; Ross, I. G.; Moomaw, W. R. J. Mol. Spectrosc. 1988, 132, 492–544. doi:10.1016/0022-2852(88)90343-8
Return to citation in text: [1] -
Suga, K.; Kinoshita, M. Bull. Chem. Soc. Jpn. 1982, 55, 1695–1704. doi:10.1246/bcsj.55.1695
Return to citation in text: [1] -
Baytekin, B.; Baytekin, H. T.; Schalley, C. Org. Biomol. Chem. 2006, 4, 2825–2841. doi:10.1039/b604265a
Return to citation in text: [1] -
Hay, B. P.; Bryantsev, V. S. Chem. Commun. 2008, 2417–2428. doi:10.1039/b800055g
Return to citation in text: [1] -
Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballister, P.; Costa, A.; Deyà, P. M. Angew. Chem., Int. Ed. 2002, 41, 3389–3392. doi:10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S
Return to citation in text: [1] -
Schaller, T.; Büchele, U. P.; Klärner, F.-G.; Bläser, D.; Boese, R.; Brown, S. P.; Spiess, H. W.; Koziol, F.; Kussmann, J.; Ochsenfeld, C. J. Am. Chem. Soc. 2007, 129, 1293–1303. doi:10.1021/ja0666351
Return to citation in text: [1] -
Parac, M.; Etinski, M.; Peric, M.; Grimme, S. J. Chem. Theory Comput. 2005, 1, 1110–1118. doi:10.1021/ct050122n
Return to citation in text: [1] -
Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O’Neill, D. P.; DiStasio, R. C., Jr.; Lochan, R. A.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Van Voorhis, T.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C.-P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W. Z.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock, H. L., III; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. J.; Warshel, A.; Hehre, W. J.; Schaefer, H. F.; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. doi:10.1039/b517914a
Return to citation in text: [1]
| 1. | Chen, C.-W.; Whitlock, H. W., Jr. J. Am. Chem. Soc. 1978, 100, 4921–4922. doi:10.1021/ja00483a063 |
| 2. | Zimmerman, S. C. Top. Curr. Chem. 1993, 165, 71–102. doi:10.1007/BFb0111281 |
| 3. | Klärner, F.-G.; Kahlert, B. Acc. Chem. Res. 2003, 36, 919–932. doi:10.1021/ar0200448 |
| 4. | Harmata, M. Acc. Chem. Res. 2004, 37, 862–873. doi:10.1021/ar030164v |
| 5. | Rowan, A. E.; Elemans, J. A. A. W.; Nolte, R. J. M. Acc. Chem. Res. 1999, 32, 995–1006. doi:10.1021/ar9702684 |
| 6. | Katz, J. L.; Geller, B. J.; Foser, P. D. Chem. Commun. 2007, 1026–1028. doi:10.1039/b615336d |
| 7. | Wei, T.-B.; Wei, W.; Cao, C.; Zhang, Y.-M. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 1218–1228. doi:10.1080/10426500701613154 |
| 8. | Bhattacharya, S.; Tominaga, K.; Kimura, T.; Uno, H.; Komatsu, N. Chem. Phys. Lett. 2007, 433, 395–402. doi:10.1016/j.cplett.2006.11.039 |
| 9. | Skibinski, M.; Gómez, R.; Lork, E.; Azov, V. A. Tetrahedron 2009, 65, 10348–10354. doi:10.1016/j.tet.2009.10.052 |
| 10. | Nishiuchi, T.; Kuwatani, Y.; Nishinaga, T.; Iyoda, M. Chem.–Eur. J. 2009, 15, 6838–6847. doi:10.1002/chem.200900623 |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 33. | Waluk, J.; Grabowska, A.; Pakulstroka, B. J. Lumin. 1980, 21, 277–291. doi:10.1016/0022-2313(80)90007-1 |
| 34. | Imes, K. K.; Ross, I. G.; Moomaw, W. R. J. Mol. Spectrosc. 1988, 132, 492–544. doi:10.1016/0022-2852(88)90343-8 |
| 35. | Suga, K.; Kinoshita, M. Bull. Chem. Soc. Jpn. 1982, 55, 1695–1704. doi:10.1246/bcsj.55.1695 |
| 16. | Mandado, M.; Hermida-Ramón, J. M.; Estévez, C. M. THEOCHEM 2006, 854, 1–9. |
| 17. | Hermida-Ramón, J. M.; Estévez, C. M. Chem.–Eur. J. 2007, 13, 4743–4749. doi:10.1002/chem.200601836 |
| 18. | Hermida-Ramón, J. M.; Mandado, M.; Sánchez-Lozano, M.; Estévez, C. M. Phys. Chem. Chem. Phys. 2010, 12, 164–169. doi:10.1039/b915483c |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 14. | Korenaga, T.; Kosaki, T.; Kawauchi, Y.; Ema, T.; Sakai, T. J. Fluorine Chem. 2006, 127, 604–609. doi:10.1016/j.jfluchem.2005.12.007 |
| 15. | Korenaga, T.; Kawauchi, Y.; Kosaki, T.; Ema, T.; Sakai, T. Bull. Chem. Soc. Jpn. 2005, 78, 2175–2179. doi:10.1246/bcsj.78.2175 |
| 29. | Azizian, J.; Karimi, A. R.; Kazemizadeh, Z.; Mohammadi, A. A.; Mohammadizadeh, M. R. J. Org. Chem. 2005, 70, 1471–1473. doi:10.1021/jo0486692 |
| 30. | Yadav, J. S.; Reddy, B. V. S.; Rao, Y. G.; Narasaiah, A. V. Chem. Lett. 2008, 37, 348–349. doi:10.1246/cl.2008.348 |
| 31. | Sangshetti, J. N.; Kokare, N. D.; Shinde, D. B. Russ. J. Org. Chem. 2009, 45, 1116–1118. doi:10.1134/S1070428009070240 |
| 32. | Zhao, Z.; Wisnoski, D.; Wolkenberg, S. E.; Leister, W. H.; Wang, Y.; Lindsley, C. W. Tetrahedron Lett. 2004, 45, 4873–4876. doi:10.1016/j.tetlet.2004.04.144 |
| 11. | Hay, B. P.; Custelcean, R. Cryst. Growth Des. 2009, 9, 2593–2545. doi:10.1021/cg900308b |
| 12. | Schottel, B. L.; Chifotides, H. T.; Dunbar, K. R. Chem. Soc. Rev. 2008, 37, 68–83. doi:10.1039/b614208g |
| 13. | Fujita, M.; Nagao, S.; Iida, M.; Ogata, K.; Ogura, K. J. Am. Chem. Soc. 1993, 115, 1574–1576. doi:10.1021/ja00057a052 |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 24. | Arotsky, J.; Butler, R.; Darby, A. C. J. Chem. Soc. C 1970, 1480–1485. doi:10.1039/J39700001480 |
| 25. | Heaton, M.; Hill, P.; Drakesmith, F. J. Fluorine Chem. 1997, 81, 133–138. doi:10.1016/S0022-1139(96)03517-8 |
| 21. | Khan, F. A.; Dash, J.; Sudheer, C.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
| 22. | Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424–2425. doi:10.1021/ja017371f |
| 23. | Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558–9559. doi:10.1021/ja001956c |
| 21. | Khan, F. A.; Dash, J.; Sudheer, C.; Sahu, N.; Parasuraman, K. J. Org. Chem. 2005, 70, 7565–7577. doi:10.1021/jo0507385 |
| 22. | Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424–2425. doi:10.1021/ja017371f |
| 23. | Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558–9559. doi:10.1021/ja001956c |
| 20. | Chou, T.-C.; Yang, M.-S.; Lin, C.-T. J. Org. Chem. 1994, 59, 661–663. doi:10.1021/jo00082a028 |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 26. | Garcia, J. G.; Fronczek, F. R.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3289–3292. doi:10.1016/S0040-4039(00)92688-1 |
| 27. | Garcia, J. G.; McLaughlin, M. L. Tetrahedron Lett. 1991, 32, 3293–3296. doi:10.1016/S0040-4039(00)92689-3 |
| 1. | Chen, C.-W.; Whitlock, H. W., Jr. J. Am. Chem. Soc. 1978, 100, 4921–4922. doi:10.1021/ja00483a063 |
| 2. | Zimmerman, S. C. Top. Curr. Chem. 1993, 165, 71–102. doi:10.1007/BFb0111281 |
| 3. | Klärner, F.-G.; Kahlert, B. Acc. Chem. Res. 2003, 36, 919–932. doi:10.1021/ar0200448 |
| 4. | Harmata, M. Acc. Chem. Res. 2004, 37, 862–873. doi:10.1021/ar030164v |
| 5. | Rowan, A. E.; Elemans, J. A. A. W.; Nolte, R. J. M. Acc. Chem. Res. 1999, 32, 995–1006. doi:10.1021/ar9702684 |
| 6. | Katz, J. L.; Geller, B. J.; Foser, P. D. Chem. Commun. 2007, 1026–1028. doi:10.1039/b615336d |
| 7. | Wei, T.-B.; Wei, W.; Cao, C.; Zhang, Y.-M. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 1218–1228. doi:10.1080/10426500701613154 |
| 8. | Bhattacharya, S.; Tominaga, K.; Kimura, T.; Uno, H.; Komatsu, N. Chem. Phys. Lett. 2007, 433, 395–402. doi:10.1016/j.cplett.2006.11.039 |
| 9. | Skibinski, M.; Gómez, R.; Lork, E.; Azov, V. A. Tetrahedron 2009, 65, 10348–10354. doi:10.1016/j.tet.2009.10.052 |
| 10. | Nishiuchi, T.; Kuwatani, Y.; Nishinaga, T.; Iyoda, M. Chem.–Eur. J. 2009, 15, 6838–6847. doi:10.1002/chem.200900623 |
| 36. | Baytekin, B.; Baytekin, H. T.; Schalley, C. Org. Biomol. Chem. 2006, 4, 2825–2841. doi:10.1039/b604265a |
| 15. | Korenaga, T.; Kawauchi, Y.; Kosaki, T.; Ema, T.; Sakai, T. Bull. Chem. Soc. Jpn. 2005, 78, 2175–2179. doi:10.1246/bcsj.78.2175 |
| 41. | Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O’Neill, D. P.; DiStasio, R. C., Jr.; Lochan, R. A.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Van Voorhis, T.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C.-P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W. Z.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock, H. L., III; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. J.; Warshel, A.; Hehre, W. J.; Schaefer, H. F.; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. doi:10.1039/b517914a |
| 15. | Korenaga, T.; Kawauchi, Y.; Kosaki, T.; Ema, T.; Sakai, T. Bull. Chem. Soc. Jpn. 2005, 78, 2175–2179. doi:10.1246/bcsj.78.2175 |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 39. | Schaller, T.; Büchele, U. P.; Klärner, F.-G.; Bläser, D.; Boese, R.; Brown, S. P.; Spiess, H. W.; Koziol, F.; Kussmann, J.; Ochsenfeld, C. J. Am. Chem. Soc. 2007, 129, 1293–1303. doi:10.1021/ja0666351 |
| 40. | Parac, M.; Etinski, M.; Peric, M.; Grimme, S. J. Chem. Theory Comput. 2005, 1, 1110–1118. doi:10.1021/ct050122n |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
| 37. | Hay, B. P.; Bryantsev, V. S. Chem. Commun. 2008, 2417–2428. doi:10.1039/b800055g |
| 38. | Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballister, P.; Costa, A.; Deyà, P. M. Angew. Chem., Int. Ed. 2002, 41, 3389–3392. doi:10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S |
| 19. | Chou, T.-C.; Liao, K.-C.; Lin, J.-J. Org. Lett. 2005, 7, 4843–4846. doi:10.1021/ol051707z |
© 2010 Etzkorn et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
