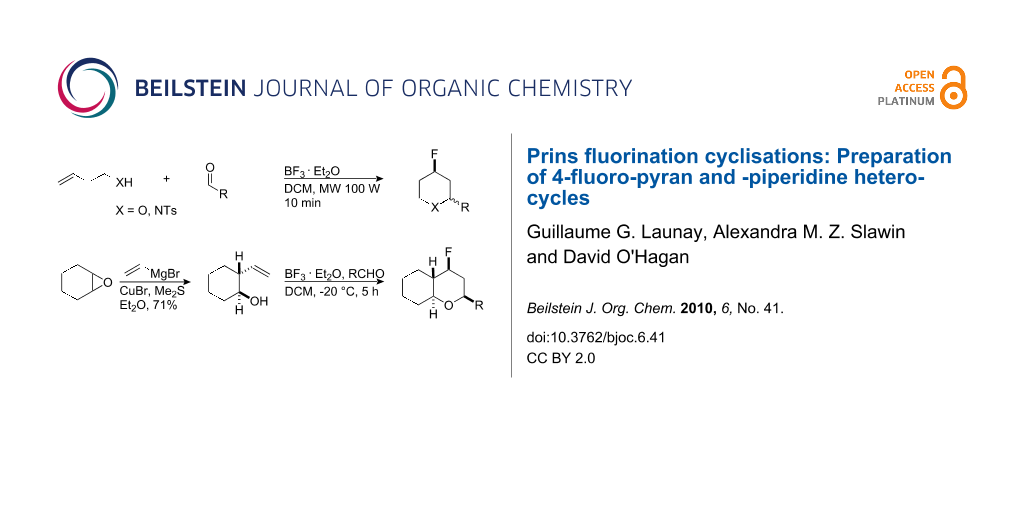
Abstract
The Prins reaction was investigated using BF3·OEt2 as a Lewis acid. It has been recently demonstrated, that if BF3·OEt2 is used in stoichiometric amounts then these reactions generate fluorinated products where the BF3·OEt2 contributes fluoride ion to quench the intermediate carbocations. In this study oxa- and aza-Prins reactions for the synthesis of 4-fluoro-pyrans and -piperidines were investigated. The products were obtained in good yields, but only with moderate diastereoselectivity. These Prins fluorination reactions can be accelerated under microwave conditions. The study extends the Prins fluorination methodology for the generation of the C–F bond in heterocycles.

Graphical Abstract
Introduction
Selective incorporation of the C–F bond into organic molecules can impart useful and attractive properties to performance materials [1-3]. To this end there are a useful but relatively limited range of fluorination reagents and methodologies available to synthetic organic chemistry, and novel methods for introducing fluorine into organic molecules continue to be valuable [4]. In this paper we focus on extending the scope of the Prins fluorination reaction as a synthetic methodology. The Prins reaction is a well established strategy for the synthesis of pyrans [5-7]. This cyclisation reaction, which occurs between a homoallylic alcohol and an aldehyde, is generally promoted by a Lewis acid. When BF3·OEt2 is used as the Lewis acid, then fluoride ion from the reagent can become incorporated into the product generating a C–F bond and a new stereogenic centre. Liberation of fluoride ion from BF3·OEt2 has, for example, been observed in epoxide ring opening reactions [8,9]. This was first recognised in a Prins reaction as an unexpected side reaction by Al-Mutairi et al. [10,11] and was noted separately by Jaber et al. [12] and subsequently by Kataoka et al. [13]. For example, homoallylic alcohol 1 was converted to pyran 2 with a high diastereoselectivity (Scheme 1) [12]. Most recently, oxa-, aza- and thia-Prins fluorination cyclisations have been carried out using ionic liquid hydrogen fluoride salts (Et4NF·5HF) as the reaction medium, without the requirement for BF3·OEt2 [14,15]. These reactions with fluoride follow from the much more commonly observed Prins reactions of halides (Cl−, Br− and I−) other than fluoride in the quenching of the intermediate oxonium intermediate [16-22].
We have explored C–F bond formation by the BF3·OEt2/Prins reaction further. In this paper we report that a wide range of 4-fluorotetrahydropyrans can be prepared by reaction of homoallylic alcohols with different aldehydes with BF3·OEt2 as the fluoride source. This Prins methodology was extended to the aza-Prins reaction using N-tosyl-homoallylic amines to generate the corresponding 4-fluoropyrrolidines. In general, these reactions leading to both the 4-fluorotetrahydropyrans and 4-fluoro-pyrrolidines occur with good to high conversions, however the diastereoselectivities are modest, particularly in the aza-Prins cases. This study also demonstrates that the conversions and reaction times can be improved using microwave conditions.
![[1860-5397-6-41-i1]](/bjoc/content/inline/1860-5397-6-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The C–F bond forming Prins reaction leading to 4-fluoropyrans [10].
Scheme 1: The C–F bond forming Prins reaction leading to 4-fluoropyrans [10].
Results and Discussion
The oxa-Prins fluorination reaction: Oxa-Prins fluorination reactions were investigated with but-3-en-1-ol (3) and a range of substituted benzaldehydes 4. Electron withdrawing groups on the aromatic ring led to the more efficient reactions (Table 1, entries a–g) to generate 4-fluoropyrans 5 with conversions of 65–73%. Diastereoselectivities were however, modest with d.r’s of between 1.9 : 1 and 5.4 : 1. With electron donating groups on the aromatic ring (Table 1, entries h–j), the reactions were inefficient and conversions dropped dramatically. The saturated aliphatic aldehyde, hexanal (entry k), resulted in a good conversion, although the corresponding 4-fluoropyran products were obtained with poor diastereoselectivity (2 : 1). In the case of 2-methylcinnamaldehyde (entry l), the conversion was poor.
Table 1: Prins fluorination reaction of homoallylic alcohol (3) with various aldehydes 4 giving substituted fluoropyrans 5. Reaction conditions: BF3·OEt2 (1 equiv), but-3-en-1-ol (1 equiv), aldehyde (1 equiv), DCM, rt, 2 h.
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-41-i4.svg?max-width=637&scale=1.0)
|
|||
| pyran | aldehyde | conversion | d.r. (syn/anti) |
|---|---|---|---|
| 5a | 4-nitrobenzaldehyde | 67% | 1.9/1 |
| 5b | 2-fluorobenzaldehyde | 66% | 4.5/1 |
| 5c | 3-fluorobenzaldehyde | 66% | 3.4/1 |
| 5d | 4-fluorobenzaldehyde | 66% | 4.5/1 |
| 5e | 2-bromobenzaldehyde | 65% | 5.4/1 |
| 5f | 3-bromobenzaldehyde | 73% | 3.8/1 |
| 5g | 4-bromobenzaldehyde | 90% | 4.8/1 |
| 5h | 2-methoxybenzaldehyde | <5% | 1.3/1 |
| 5i | 4-methoxybenzaldehyde | 20% | 2.4/1 |
| 5j |
2,3,6-trimethoxybenz-
aldehyde |
no reaction | / |
| 5k | hexanal | 76% | 2/1 |
| 5l | 2-methylcinnamaldehyde | <5% | / |
Microwave – oxa-Prins: The Prins fluorination reactions were then investigated under microwave conditions (Table 2 and Scheme 3). Reaction times were significantly reduced to 10 min and, in general, the conversions were higher than under the more classical conditions. The diastereoselectivity appears to decrease a little, and in some cases there is an inversion in the major diastereoisomer, e.g., with 4-methoxybenzaldehyde.
Table 2: Prins fluorination microwave (100 W, 50 °C, 10 min) reactions using homoallylic alcohol (3), an aldehyde and BF3·OEt2 in DCM.
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-41-i5.svg?max-width=637&scale=1.0)
|
|||
| entry | aldehyde | d.r. (5/6) | conversion |
|---|---|---|---|
| 5a | 4-nitrobenzaldehyde | 1.5/1 | 53% |
| 5e | 2-bromobenzaldehyde | 3/1 | 92% |
| 5f | 3-bromobenzaldehyde | 1.8/1 | 93% |
| 5g | 4-bromobenzaldehyde | 2.3/1 | 83% |
| 5i | 4-methoxybenzaldehyde | 1/1.2 | 41% |
| 5k | hexanal | 1.8/1 | 91% |
| 5m | benzaldehyde | 3.4/1 | 66% |
A series of low temperature studies was carried out in an attempt to improve the diastereoselectivity. When the temperature was lowered to −20 °C (Table 3) the diastereoselectivity increased from ~2/1 to 10/1 and generally in good yields, but with a significant increase in the reaction time. Lowering the temperature below −20 °C did not lead to a significant improvement.
In order to confirm the configuration of the major diastereoisomer, an X-ray structure analysis was carried out on the major diastereoisomer produced in the low temperature reaction between alcohol 3 and 4-nitrobenzaldehyde. The X-ray confirmed that the major diastereoisomer is syn-5a as shown in Figure 1.
![[1860-5397-6-41-1]](/bjoc/content/figures/1860-5397-6-41-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of syn-5a.
Figure 1: X-ray crystal structure of syn-5a.
In order to elaborate one of the Prins fluorination products, pyran 5m was subjected to hydrogenolysis [23] as illustrated in Scheme 2. This resulted in the efficient conversion to the corresponding open chain compound 3-fluoro-5-phenylpentyl acetate (7).
![[1860-5397-6-41-i2]](/bjoc/content/inline/1860-5397-6-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ring opening hydrogenation of an oxa-Prins product.
Scheme 2: Ring opening hydrogenation of an oxa-Prins product.
The structural diversity of the Prins fluorination reaction was extended using 2-vinylcyclohexan-ol (8) as a substrate. Vinylcyclohexanol 8 was prepared by treatment of cyclohexene oxide (10) with vinylmagnesium bromide in the presence of CuBr/DMSO as illustrated in Table 4 [24]. This gave a single diastereoisomer of 8 which was used in the Prins fluorination reactions.
Prins fluorination reactions, at −20 °C, with cyclohexanol 8 using benzaldehyde and 4-nitrobenzaldehyde gave rise to the corresponding bicyclic products 9 with good diastereoselectivity (10/1) and in moderate yields (Table 4).
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-41-i6.svg?max-width=637&scale=1.0)
The X-ray crystal structure of the predominant bicyclic diastereoisomer was determined and the syn-stereoisomer 9b, as shown in Figure 2, was confirmed as the major product of this Prins reaction.
![[1860-5397-6-41-2]](/bjoc/content/figures/1860-5397-6-41-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of the major bicyclic tetrahydropyran diastereoisomer 9b.
Figure 2: X-ray crystal structure of the major bicyclic tetrahydropyran diastereoisomer 9b.
It is interesting to note that when (E)- and (Z)-hex-3-en-1-ol (11a) and (11b) were used as substrates, the double bond stereochemistry is retained. Only two diastereoisomers were observed in the products, differing only in the orientation of the fluorine. In general, these reactions gave good diastereoselectivities in moderate to good yields (Scheme 3).
![[1860-5397-6-41-i3]](/bjoc/content/inline/1860-5397-6-41-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction using (E)-11a and (Z)-11b hex-3-en-1-ols with 4-nitrobenzaldehyde to generate 4-fluorotetrahydropyrans 12.
Scheme 3: Reaction using (E)-11a and (Z)-11b hex-3-en-1-ols with 4-nitrobenzaldehyde to generate 4-fluorotetr...
Aza-Prins fluorination reaction
The aza-Prins reaction is less well known but has been exploited, e.g., in tandem reactions in alkaloid synthesis [25,26]. Recently, it has been used to incorporate a halogen atom at the 4 position of a piperidine ring as reported by Carballo et al. [16], and most recently an example incorporating fluorine at the 4-position of a piperidine using Et4NF·5HF has been reported [14,15]. We have extended this study to explore more fully the aza-Prins fluorination reaction of homoallylamine 13. Several amine protecting groups such as benzoyl and Boc were examined, but only the N-tosyl homoallylamine 13 proved to be a useful substrate as illustrated in Table 5. The tosylamine 13 was prepared (41% yield) by the reaction of 1-bromobut-3-ene with N-tosylamine in the presence of potassium carbonate. The yields and diastereoselectivity of the aza-Prins reactions with a variety of aldehydes were comparable to the oxa-Prins reactions, but longer reaction times were required (typically 36 h). The reactions are summarised in Table 5. In contrast to the oxa-Prins reactions, lowering the temperature of the reaction had no measurable influence on the diastereoselectivity (Table 5, entry 4a).
Table 5: Aza-Prins reaction between N-homoallyl-N-tosylamine (13) and aldehydes 14 in the presence of BF3·OEt2. Reaction conditions (except entry 2): Amine 13 (1 equiv), aldehyde (1 equiv), BF3·OEt2 for 36 h at rt; yield refers to the isolation of both stereoisomers in each case except for entry 14j and 14k where the yields refer to the isolation of diastereoisomer mixture.
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-41-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | aldehyde | (syn/anti 14) | conversion |
|---|---|---|---|
| 14a | 4-nitrobenzaldehyde | 1/1 | 65% |
| 14aa | 4-nitrobenzaldehyde | 1/1 | 71% |
| 14b | hexanal | 2/1 | 75% |
| 14c | 4-bromobenzaldehyde | 1/1 | 71% |
| 14d | 3-bromobenzaldehyde | 2/1 | 61% |
| 14e | 2-bromobenzaldehyde | 1/1.6 | 50% |
| 14f | 4-fluorobenzaldehyde | 1.8/1 | 70% |
| 14g | 3-fluorobenzaldehyde | 2/1 | 67% |
| 14h | 4-methoxybenzaldehyde | 2.5/1 | 23% |
| 14i | 2-methylcinnamaldehyde | no reaction | – |
| 14j | acetaldehyde | 1/1 | 73% |
| 14k | isobutyraldehyde | 1/1 | 82% |
aLow temperature reaction (−20 °C, 48 h)
All the aldehydes used in the study (Table 5) gave universally poor diastereoselectivities, however the conversions were generally good except in the case of where a relatively strong electron donating group was present on the aromatic ring (e.g. 14h, Table 5). There was no reaction with the α,β-unsaturated aldehyde, 2-methylcinnamaldehyde (14i). In the case of aliphatic aldehydes (14b, 14j, 14k), the conversions were high, but again the diastereoselectivity was poor.
Microwave – aza-Prins: Following the observation of reduced reaction times in the oxa-Prins under microwave conditions, it was of interest to investigate the aza-Prins fluorination reaction under microwave conditions, particularly as these reactions were much slower.
In the event, the microwave reactions proved to be very efficient giving products with improved conversions after 30 min as shown in Table 6. Despite this improvement in rate, there was no significant improvement in the diastereoselectivity of the reaction products. This should be contrasted with the study using an HF containing ionic liquid (Et4NF·5HF) in place of BF3·OEt2, where diastereoselectivities for similar aza-Prins reactions where around 7:1 to 10:1 in favour of the syn-products [14,15].
Table 6: aza-Prins reaction under microwave conditions. Reaction conditions: amine 13 (1 equiv), aldehyde (1 equiv), BF3·OEt2, microwave 100 W, 50 °C, 30 min.
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-41-i8.svg?max-width=637&scale=1.0)
|
|||
| entry | aldehyde | syn/anti | conversion |
|---|---|---|---|
| 14a | 4-nitrobenzaldehyde | 1.3/1 | 61% |
| 14b | hexanal | 1.9/1 | 83% |
| 14c | 4-bromobenzaldehyde | 1.9/1 | 68% |
| 14f | 4-fluorobenzaldehyde | 1.5/1 | 63% |
| 14i | acetaldehyde | 1.2/1 | 77% |
| 14k | isobutyraldehyde | 1.9/1 | 83% |
The crystal structure of the minor anti-diastereoisomer of the piperidine product 14d was determined and is shown in Figure 3.
![[1860-5397-6-41-3]](/bjoc/content/figures/1860-5397-6-41-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of the minor anti-piperidine product 14d.
Figure 3: X-ray crystal structure of the minor anti-piperidine product 14d.
Conclusion
Selective methods for fluorination are finding increasing utility in pharmaceutical, agrochemicals and fine chemicals research. The BF3·OEt mediated oxa- and aza-Prins fluorination extends the methodologies available for the synthesis of C–F bonds, particularly concomitant with O- and N-heterocycle assembly. In general the diastereoselectivities are poor in these reactions, however they can be improved in the oxa-Prins case by lowering the temperature of the reactions to −20 °C. Both these oxa- and aza-Prins reactions can be significantly accelerated under microwave conditions.
Supporting Information
| Supporting Information File 1: Experimental and characterisation details of synthesised compounds. | ||
| Format: PDF | Size: 449.2 KB | Download |
References
-
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
O'Hagan, D.; Hunter, L.; Kirsch, P.; Slawin, A. M. Z. Angew. Chem., Int. Ed. 2009, 48, 5457–5460. doi:10.1002/anie.200901956
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell Publishing Ltd.: Oxford, 2006.
Return to citation in text: [1] -
Yadav, J. S.; Subba Reddy, B. V.; Gupta, M. K.; Biswas, S. K. Synthesis 2004, 2711–2715. doi:10.1055/s-2004-831220
Return to citation in text: [1] -
Arundale, E.; Mikeska, L. A. Chem. Rev. 1952, 51, 505–555. doi:10.1021/cr60160a004
Return to citation in text: [1] -
Alder, R. W.; Harvey, J. N.; Oakley, M. T. J. Am. Chem. Soc. 2002, 124, 4960–4961. doi:10.1021/ja025902+
Return to citation in text: [1] -
Ashwell, M.; Jackson, R. F. W.; Kirk, J. M. Tetrahedron 1990, 46, 7429–7442. doi:10.1016/S0040-4020(01)89058-X
Return to citation in text: [1] -
Ashwell, M.; Jackson, R. F. W. J. Chem. Soc., Chem. Commun. 1988, 282–283.
Return to citation in text: [1] -
Al-Mutairi, E. H.; Crosby, S. R.; Darzi, J.; Harding, J. R.; Hughes, R. A.; King, C. D.; Simpson, T. J.; Smith, R. W.; Willis, C. L. Chem. Commun. 2001, 835–836. doi:10.1039/b101414p
Return to citation in text: [1] [2] -
Crosby, S. R.; Harding, J. R.; King, C. D.; Parker, G. D.; Willis, C. L. Org. Lett. 2002, 4, 577–580. doi:10.1021/ol0102850
Return to citation in text: [1] -
Jaber, J. J.; Mitsui, K.; Rychnovsky, S. D. J. Org. Chem. 2001, 66, 4679–4686. doi:10.1021/jo010232w
Return to citation in text: [1] [2] -
Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054
Return to citation in text: [1] -
Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c
Return to citation in text: [1] [2] [3] -
Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 1, 103–109. doi:10.1002/ejoc.200800872
Return to citation in text: [1] [2] [3] -
Carballo, R. M.; Ramírez, M. A.; Rodríguez, M. L.; Martin, V. S.; Padrón, J. I. Org. Lett. 2006, 8, 3837–3840. doi:10.1021/ol061448t
Return to citation in text: [1] [2] -
Miranda, P. O.; Carballo, R. M.; Ramírez, M. A.; Martin, V. S.; Padrón, J. I. ARKIVOC 2007, iv, 331–343.
Return to citation in text: [1] -
Yadava, J. S.; Reddy, B. V. S.; Chaya, D. N.; Kumara, G. G. K. S. N.; Naresh, P.; Jagadeesh, B. Tetrahedron Lett. 2009, 50, 1799–1802. doi:10.1016/j.tetlet.2009.01.148
Return to citation in text: [1] -
Yadava, J. S.; Reddy, B. V. S.; Chaya, D. N.; Kumara, G. G. K. S. N.; Arivind, S.; Kunwar, A. C.; Madavi, C. Tetrahedron Lett. 2008, 49, 3330–3334. doi:10.1016/j.tetlet.2008.03.006
Return to citation in text: [1] -
Dobbs, A. P.; Pivnevi, L.; Penny, M. J.; Martinovic, S.; Lley, J. N.; Stephenson, P. T. Chem. Commun. 2006, 3134–3136. doi:10.1039/b606121d
Return to citation in text: [1] -
Dobbs, A. P.; Guesné, S. J. J.; Martinović, S.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 2003, 68, 7880–7883. doi:10.1021/jo034981k
Return to citation in text: [1] -
Silva, L. F., Jr.; Quintiliano, S. A. Tetrahedron Lett. 2009, 50, 2256–2260. doi:10.1016/j.tetlet.2009.02.202
Return to citation in text: [1] -
Baker, R. H.; Cornell, K. H.; Cron, M. J. J. Am. Chem. Soc. 1948, 70, 1490–1492. doi:10.1021/ja01184a057
Return to citation in text: [1] -
Tobia, D.; Rickborn, B. J. Org. Chem. 1989, 54, 777–782. doi:10.1021/jo00265a013
Return to citation in text: [1] -
Armstrong, A.; Bhonoah, Y.; Shanahan, S. E. J. Org. Chem. 2007, 72, 8019–8024. doi:10.1021/jo701536a
Return to citation in text: [1] -
Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335–1338. doi:10.1021/ol0501267
Return to citation in text: [1]
| 14. | Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c |
| 15. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 1, 103–109. doi:10.1002/ejoc.200800872 |
| 1. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 2. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | O'Hagan, D.; Hunter, L.; Kirsch, P.; Slawin, A. M. Z. Angew. Chem., Int. Ed. 2009, 48, 5457–5460. doi:10.1002/anie.200901956 |
| 10. | Al-Mutairi, E. H.; Crosby, S. R.; Darzi, J.; Harding, J. R.; Hughes, R. A.; King, C. D.; Simpson, T. J.; Smith, R. W.; Willis, C. L. Chem. Commun. 2001, 835–836. doi:10.1039/b101414p |
| 11. | Crosby, S. R.; Harding, J. R.; King, C. D.; Parker, G. D.; Willis, C. L. Org. Lett. 2002, 4, 577–580. doi:10.1021/ol0102850 |
| 16. | Carballo, R. M.; Ramírez, M. A.; Rodríguez, M. L.; Martin, V. S.; Padrón, J. I. Org. Lett. 2006, 8, 3837–3840. doi:10.1021/ol061448t |
| 8. | Ashwell, M.; Jackson, R. F. W.; Kirk, J. M. Tetrahedron 1990, 46, 7429–7442. doi:10.1016/S0040-4020(01)89058-X |
| 9. | Ashwell, M.; Jackson, R. F. W. J. Chem. Soc., Chem. Commun. 1988, 282–283. |
| 14. | Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c |
| 15. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 1, 103–109. doi:10.1002/ejoc.200800872 |
| 5. | Yadav, J. S.; Subba Reddy, B. V.; Gupta, M. K.; Biswas, S. K. Synthesis 2004, 2711–2715. doi:10.1055/s-2004-831220 |
| 6. | Arundale, E.; Mikeska, L. A. Chem. Rev. 1952, 51, 505–555. doi:10.1021/cr60160a004 |
| 7. | Alder, R. W.; Harvey, J. N.; Oakley, M. T. J. Am. Chem. Soc. 2002, 124, 4960–4961. doi:10.1021/ja025902+ |
| 24. | Tobia, D.; Rickborn, B. J. Org. Chem. 1989, 54, 777–782. doi:10.1021/jo00265a013 |
| 25. | Armstrong, A.; Bhonoah, Y.; Shanahan, S. E. J. Org. Chem. 2007, 72, 8019–8024. doi:10.1021/jo701536a |
| 26. | Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335–1338. doi:10.1021/ol0501267 |
| 14. | Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c |
| 15. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 1, 103–109. doi:10.1002/ejoc.200800872 |
| 10. | Al-Mutairi, E. H.; Crosby, S. R.; Darzi, J.; Harding, J. R.; Hughes, R. A.; King, C. D.; Simpson, T. J.; Smith, R. W.; Willis, C. L. Chem. Commun. 2001, 835–836. doi:10.1039/b101414p |
| 12. | Jaber, J. J.; Mitsui, K.; Rychnovsky, S. D. J. Org. Chem. 2001, 66, 4679–4686. doi:10.1021/jo010232w |
| 23. | Baker, R. H.; Cornell, K. H.; Cron, M. J. J. Am. Chem. Soc. 1948, 70, 1490–1492. doi:10.1021/ja01184a057 |
| 13. | Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054 |
| 12. | Jaber, J. J.; Mitsui, K.; Rychnovsky, S. D. J. Org. Chem. 2001, 66, 4679–4686. doi:10.1021/jo010232w |
| 16. | Carballo, R. M.; Ramírez, M. A.; Rodríguez, M. L.; Martin, V. S.; Padrón, J. I. Org. Lett. 2006, 8, 3837–3840. doi:10.1021/ol061448t |
| 17. | Miranda, P. O.; Carballo, R. M.; Ramírez, M. A.; Martin, V. S.; Padrón, J. I. ARKIVOC 2007, iv, 331–343. |
| 18. | Yadava, J. S.; Reddy, B. V. S.; Chaya, D. N.; Kumara, G. G. K. S. N.; Naresh, P.; Jagadeesh, B. Tetrahedron Lett. 2009, 50, 1799–1802. doi:10.1016/j.tetlet.2009.01.148 |
| 19. | Yadava, J. S.; Reddy, B. V. S.; Chaya, D. N.; Kumara, G. G. K. S. N.; Arivind, S.; Kunwar, A. C.; Madavi, C. Tetrahedron Lett. 2008, 49, 3330–3334. doi:10.1016/j.tetlet.2008.03.006 |
| 20. | Dobbs, A. P.; Pivnevi, L.; Penny, M. J.; Martinovic, S.; Lley, J. N.; Stephenson, P. T. Chem. Commun. 2006, 3134–3136. doi:10.1039/b606121d |
| 21. | Dobbs, A. P.; Guesné, S. J. J.; Martinović, S.; Coles, S. J.; Hursthouse, M. B. J. Org. Chem. 2003, 68, 7880–7883. doi:10.1021/jo034981k |
| 22. | Silva, L. F., Jr.; Quintiliano, S. A. Tetrahedron Lett. 2009, 50, 2256–2260. doi:10.1016/j.tetlet.2009.02.202 |
© 2010 Launay et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)