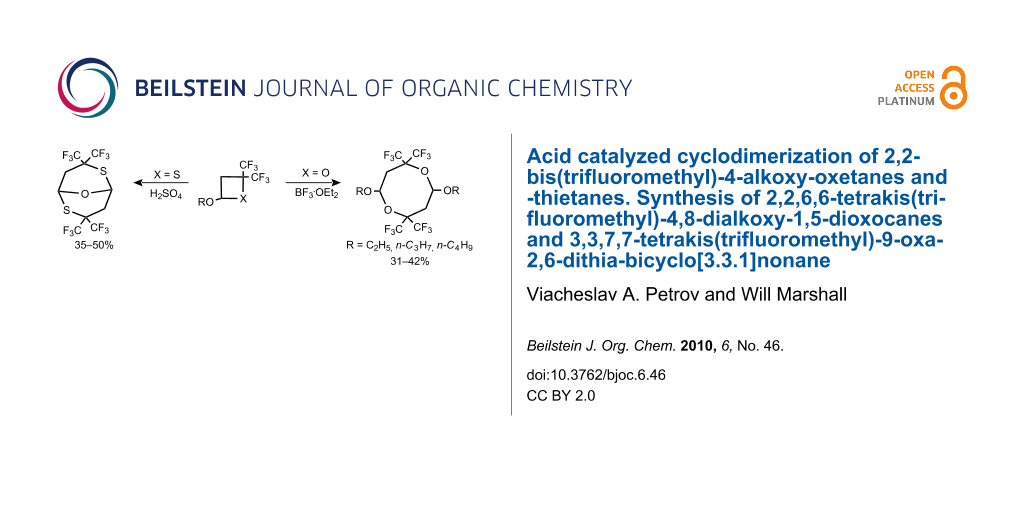
Abstract
Treatment of 2,2-bis(trifluoromethyl)-4-R-oxetanes (R = C2H5O, n-C3H7O, n-C4H9O) with BF3·OEt2 in CH2Cl2 solvent results in spontaneous electrophilic [4 + 4] cyclodimerization with the formation of the corresponding 2,2,6,6-tetrakis(trifluoromethyl)-4,8-dialkoxy-1,5-dioxocanes, isolated in 31–42% yield. The structures of two products (R = C2H5O and n-C3H7O) were established by single crystal X-ray diffraction. The corresponding oxetane carrying the bulky t-C4H9O group has different reactivity towards BF3·OEt2, slowly producing a mixture of two acyclic, unsaturated products.
Clean and spontaneous reaction with alcohols is another interesting transformation of oxetanes described in this paper. The reaction leads to high yield formation of the corresponding acetals (CF3)2C(OH)CH2CH(OR)OR′.
Structurally related 2,2-bis(trifluoromethyl)-4-R-thietanes (R = i-C3H7O, t-C4H9O and C2H5O) have different reactivity towards electrophiles. They are totally inert to the action of BF3·OEt2 and rapidly react with a protic acid (H2SO4) forming the same product, 3,3,7,7-tetrakis(trifluoromethyl)-9-oxa-2,6-dithia-bicyclo[3.3.1]nonane in 35–50% yield. The structure of this product was established by single crystal X-ray diffraction.

Graphical Abstract
Introduction
Polyfluorinated 2,2-bis(trifluoromethyl)-4-alkoxy-oxetanes and -thietanes are readily available materials, prepared by [2 + 2] cycloaddition of vinyl ethers with hexafluoroacetone [1-3] or hexafluorothioacetone [4,5], respectively. Although both groups of compounds have been known for over 40 years, reports on their chemical transformations are limited. Among the reported reactions of oxetanes are hydrolysis of 2,2-bis(trifluoromethyl)-4-alkoxyoxetanes 1 leading to the formation of 4,4,4-trifluoro-3-(trifluoromethyl)-3-hydroxybutanal [1,6] and thermal or acid catalyzed isomerization of 2,2-bis(trifluoromethyl)-4-n-butoxyoxetane into (E)-4-n-butoxy-1,1,1-trifluoro-2-(trifluoromethyl)but-3-en-2-ol [2].
Some compounds containing two 2,2-bis(trifluoromethyl)oxetane units, such as bis-4,4-(trifluoromethyl)oxetan-2-yl ether, were reported to undergo Lewis acid catalyzed polymerization [3].
Known reactions of 2,2-bis(trifluoromethyl)-4-alkoxythietanes (R = CH3O and C2H5O) include the formation of 4-(4,4-bis(trifluoromethyl)thietan-2-yloxy)-2,2-bis(trifluoromethyl)thietane on treatment with H2SO4 [4], thiophilic ring opening by the action of alkyl magnesium or lithium reagents [4], the recently reported oxidation with selective formation of the corresponding S-oxides [7], and an unusual reductive ring expansion leading to the corresponding dihydrothiophenes [7].
As part of a program to identify new, readily available fluorinated monomers, we have carried out a comparative study of the reactivity of 2,2-bis(trifluoromethyl)-4-alkoxy- oxetanes and -thietanes towards acids. The results of this study are reported in this paper.
Results and Discussion
In sharp contrast to the reported isomerization of 2,2-bis(trifluoromethyl)-4-alkoxyoxetanes catalyzed by protic acids [2], the reaction of oxetanes 1a–c with a catalytic amount of Lewis acid leads to a completely different reaction course. The addition of boron trifluoride etherate catalyst to a solution of the oxetane in dichloromethane resulted in a spontaneous and mildly exothermic reaction. A very interesting feature of this process is the appearance of highly intensive blue or blue-green colour upon the addition of the first drop of the catalyst. The colour of the reaction mixture rapidly changes to dark red and finally to brown and, at this stage, usually the formation of a precipitate is observed. The solid products 2a–c were isolated in moderate yields after filtration of the cold reaction mixture and washing of the filter cake with water. Analytically pure samples were prepared by crystallization from hexane. 1H, and 13C NMR and IR spectroscopic data show the absence of a C=C bond in all of the isolated products. Due to the fact, that 1H and 19F NMR spectra of 2a, b (see Supporting Information File 1, Table 1, Entries 1, 2) and starting oxetanes 1a, b have a similar appearance, spectroscopic data were not sufficient for an unambiguous assignment of the structure. Consequently, structure assignments for 2a and 2b were made based on single crystal X-ray diffraction data (see Supporting Information File 2).
Both compounds were found to have a symmetrical 2,2,6,6-tetrakis(trifluoromethyl)-1,5-dioxocane core with a trans-orientation of two alkoxy groups located at positions 4 and 8 (Scheme 1 and Figure 1 for structure of 2a and 2b, Supporting Information File 2 for single crystal X-ray data).
![[1860-5397-6-46-i1]](/bjoc/content/inline/1860-5397-6-46-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrophilic [4 + 4] dimerization of oxetanes 1a–c under action of BF3·OEt2 catalyst.
Scheme 1: Electrophilic [4 + 4] dimerization of oxetanes 1a–c under action of BF3·OEt2 catalyst.
Since 1H, 13C and 19F NMR spectra of compound 2c were similar to the NMR spectra of 2a, b, it is assumed that compound 2c also has a 2,2,6,6-tetrakis(trifluoromethyl)-1,5-dioxocane structure.
![[1860-5397-6-46-1]](/bjoc/content/figures/1860-5397-6-46-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP drawing of compounds 2a and 2b with thermal ellipsoids drawn to the 50% probability level.
Figure 1: ORTEP drawing of compounds 2a and 2b with thermal ellipsoids drawn to the 50% probability level.
Despite the fact that yields of 1,5-dioxocanes 2a–c in the reaction of oxetanes with BF3·OEt2 are modest, the process itself is simple, reproducible and provides easy access to this new group of stable polyfluorinated 1,5-dioxocanes. It should also be pointed out, that examples of electrophilic [4 + 4] cycloaddition reactions are extremely rare and limited to two examples: the reaction of the oxetane (derived from the cycloaddition of 1,1-dimethoxyethylene and 2,2-dimethylcyclopropanone), leading to a stable hydrocarbon 1,5-dioxocane [8] and the formation of the corresponding fluorinated 1,5-dioxocane intermediate [9,10] observed in the isomerization of 2-ethoxy-4-(perfluoropropan-2-ylidene)oxetane [11,12].
The chemical behavior of oxetane 1d carrying the bulky t-C4H9O substituent is different to 1a–c. The addition of BF3·OEt2 as catalyst to a solution of 1d in CH2Cl2 is not exothermic and results only in a faint blue-greenish color in this case. In sharp contrast to the reaction of oxetanes 1a–c with BF3·OEt2, this process is rather slow (85% conversion after 1 week at 25 °C) and it leads to the formation of a mixture of olefinic products 2d and 2e (Scheme 2).
![[1860-5397-6-46-i2]](/bjoc/content/inline/1860-5397-6-46-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
A sample of pure 2d was isolated by fractional distillation of the reaction mixture under reduced pressure. The structure of the olefin 2d was established by single crystal X-ray diffraction analysis (see Supporting Information File 2).
Hydrocarbon oxetanes were reported to react with alcohols under relatively mild conditions [13]. It is interesting, that electron deficient oxetanes 1 also have similar reactivity and rapidly react with alcohols in the absence of the catalyst. The reaction leads to a ring opening with the formation of the corresponding acetals of 4,4,4-trifluoro-3-(trifluoromethyl)-3-hydroxybutanal. For example, the addition of 1b or 1c to an excess of methanol results in a fast and mildly exothermic reaction, leading to selective formation of acetals 3a or 3b, respectively (Scheme 3).
![[1860-5397-6-46-i3]](/bjoc/content/inline/1860-5397-6-46-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 2,2-bis(trifluoromethyl)-4-alkoxyoxetanes 1b, c with methanol.
Scheme 3: Reaction of 2,2-bis(trifluoromethyl)-4-alkoxyoxetanes 1b, c with methanol.
Since the vacuum distillation of 3b lead to decomposition, the isolation of similar products in an analytically pure form was not attempted. However, removal of excess alcohol after the reaction was complete by washing with water afforded products of reasonable purity (96–98%) in >95% yield.
Although kinetic measurements were not carried out in this study, it appears that the reaction time and the exothermicity of the reaction of oxetanes correlates with the acidity of the corresponding alcohol. For example, in contrast to a mildly exothermic reaction of 1b, c with methanol (pKa = 15.5 [14,15]) the interaction of 1c with more acidic CF3CH2OH (pKa = 12.4, 12.8 [15]) or (CF3)2CHOH (pKa = 9.3 [16]) is significantly more exothermic, leading to products 3c and 3d, respectively. All reactions were completed within 1–2 h at ambient temperature. On the other hand, the reaction of 1c with the less acidic (CH3)2CHOH (pKa= 17.1 [15]) was significantly slower taking >10 h for completion at ambient temperature, as monitored by 19F NMR, and led to acetal 3e (Scheme 4).
![[1860-5397-6-46-i4]](/bjoc/content/inline/1860-5397-6-46-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of oxetane 1c with alcohols.
Scheme 4: Reaction of oxetane 1c with alcohols.
A mechanism for the reaction of these fluorinated oxetanes with Lewis acids and alcohols, is presented by Scheme 5.
![[1860-5397-6-46-i5]](/bjoc/content/inline/1860-5397-6-46-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Putative mechanism for the reaction oxetanes 2a–d with BF3·OEt2 and alcohols.
Scheme 5: Putative mechanism for the reaction oxetanes 2a–d with BF3·OEt2 and alcohols.
Coordination of the Lewis acid with the oxetane ring oxygen results in the formation of stabilized zwitterion A, which probably exists in equilibrium with the starting material. The reaction of A with a second mole of oxetane then leads to the formation of zwitterion B, which can undergo intramolecular cyclization with formation of 1,5-dioxocanes 2a–c. The Lewis acid liberated in this process is free to carry out the next catalytic cycle. It should be pointed out, that recently a zwitterion similar to B was observed in the isomerization of the cycloadduct of bis(trifluoromethyl)ketene and ethyl vinyl ether [9,10].
In the case of oxetane 1d, the main channel of the reaction involves stabilization of intermediates A and B by H+ elimination, leading to the formation of olefins 2d and 2e, respectively. Such a distinct difference in the reactivity of 1d may be a result of steric hindrance of the carbocationic center in intermediates A or B, created by the bulky (CH3)3CO-group, which favors elimination and the formation of olefins 2e and 2d.
The addition of alcohols to oxetanes 1a–c probably involves the protonation of the oxetane ring oxygen atom as the first step, followed by ring opening and addition of the alkoxy anion. This mechanism agrees well with the observed order of reactivity of alcohols, with the acidic alcohols being more reactive towards the oxetane. It should be pointed out, however, that an alternative mechanism involving the “concerted” addition of the alcohol to the oxetane cannot be ruled out at this point.
Despite the structural similarity shared with oxetanes, thietanes 4a–c display a totally different reactivity (Scheme 6, Scheme 7).
For example, no reaction was detected between thietane 4a and an excess of either methanol or hexafluoroisopropanol (25 °C, 16 h, NMR) and both 4a and 4b were found to be inert towards BF3·OEt2 (25 °C, 10 h, CH2Cl2, NMR). However, the addition of compounds 4a or 4b to concentrated H2SO4 resulted in an exothermic reaction and formation of a product, which, after single crystal X-ray diffraction analysis, was shown to be 3,3,7,7-tetrakis(trifluoromethyl)-9-oxa-2,6-dithia-bicyclo[3.3.1]nonane (5) (Scheme 6, Figure 2, Supporting Information File 2).
![[1860-5397-6-46-i6]](/bjoc/content/inline/1860-5397-6-46-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of thietanes 4a, b with H2SO4 to generate 5.
Scheme 6: Reaction of thietanes 4a, b with H2SO4 to generate 5.
![[1860-5397-6-46-2]](/bjoc/content/figures/1860-5397-6-46-2.png?scale=0.8&max-width=1024&background=FFFFFF)
Figure 2: ORTEP drawing of 5 with thermal ellipsoids drawn to the 50% probability level.
Figure 2: ORTEP drawing of 5 with thermal ellipsoids drawn to the 50% probability level.
A solid product with similar melting point (91–92 °C) was observed earlier [4] in the reaction of thietanes 4 (R = CH3 and C2H5) with concentrated H2SO4. Based on a combination of 1H, 19F NMR, mass spectrometry and elemental analysis data, the structure of 4-[4,4-bis(trifluoromethyl)thietan-2-yloxy]-2,2-bis(trifluoromethyl)thietane 5a was proposed for that product [4].
In order to clarify this result, thietane 4c (R = C2H5) was treated with H2SO4 (Scheme 7) under conditions similar to those reported previously [4]. A solid product was isolated in 45% yield, which had a similar melting point and identical 1H, 19F NMR spectra to that reported for 5a (see Supporting Information File 1, Table 1, Entry 11 and footnotes f,h), but also identical to the analytical data for compound 5 prepared from oxetanes 4a and 4b.
![[1860-5397-6-46-i7]](/bjoc/content/inline/1860-5397-6-46-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Since all experimental data obtained for the reaction of thietanes 4a–c with H2SO4 in this work are consistent, it is concluded that the main product of this reaction is dithiocin 5, rather than the isomeric ether 5a, proposed in reference [4].
A possible mechanism describing the formation of compound 5 is depicted by Scheme 8.
![[1860-5397-6-46-i8]](/bjoc/content/inline/1860-5397-6-46-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Putative mechanism for the formation bicyclic ether 5.
Scheme 8: Putative mechanism for the formation bicyclic ether 5.
Protonation of the thietane sulfur of 4, followed by ring opening, results in the formation of an oxygen stabilized carbocation D. Electrophilic attack of D at the sulfur of the second thietane molecule and loss of H+ would lead to the formation of intermediate E, structurally similar to cyloadducts 2a–c. However, the process does not stop at this stage. Protonation of the alkoxy group of E, followed by the elimination of alcohol leads to the cyclic cation F, which further undergoes cyclization through intramolecular electrophilic attack on oxygen of the alkoxy group, resulting in the formation of 3,3,7,7-tetrakis(trifluoromethyl)-9-oxa-2,6-dithia-bicyclo[3.3.1]nonane (5).
Conclusion
Despite the structural similarity, 2,2-bis(trifluoromethyl)-4-alkoxy-oxetanes and thietanes have very different reactivity towards Lewis and protic acids. While the reaction of 2,2-bis(trifluoromethyl)-4-R-oxetanes (R = C2H5, n-C3H7, n-C4H9) with BF3·OEt2 results in the fast formation of the corresponding 2,2,6,6-tetrakis(trifluoromethyl)-1,5-dioxocanes 2a–c, the corresponding thietanes 4b, c (R = i-C3H7, t-C4H9) are inert towards this Lewis acid, but rapidly react with concentrated H2SO4 with the formation of bicyclic 3,3,7,7-tetrakis(trifluoromethyl)-9-oxa-2,6-dithia-bicyclo[3.3.1]nonane 5. Reinvestigation of a previously reported reaction of 4c, (R = C2H5) with H2SO4, led us to the conclusion, that the product formed in this process has the structure 3,3,7,7-tetrakis(trifluoromethyl)-9-oxa-2,6-dithia-bicyclo[3.3.1]nonane (5), rather than 4-[4,4-bis(trifluoromethyl)thietan-2-yloxy]-2,2-bis(trifluoromethyl)-thietane (5a) as proposed earlier [4].
References
-
Davis, H. R. (Minnesota Mining and Manufacturing Co.). U.S. Patent 3164610, 1965.
Return to citation in text: [1] [2] -
Gambaryan, N. P.; Simonyan, L. A.; Petrovskii, P. V. Izv. Akad. Nauk SSSR, Ser. Khim. 1967, 918–921.
Return to citation in text: [1] [2] [3] -
Braun, R. A. (du Pont de Nemours, E. I., and Co.). U.S. Patent 3417102, 1968.
Return to citation in text: [1] [2] -
Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Middleton, W. J. J. Org. Chem. 1965, 30, 1395–1398. doi:10.1021/jo01016a011
Return to citation in text: [1] -
Davis, H. R. (Minnesota Mining and Manufacturing Co.). U.S. Patent 3284509, 1966.
Return to citation in text: [1] -
Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2009, 130, 780–787. doi:10.1016/j.jfluchem.2009.06.011
Return to citation in text: [1] [2] -
Turro, N. J.; Williams, J. R. Tetrahedron Lett. 1969, 321–323. doi:10.1016/S0040-4039(01)87682-6
Return to citation in text: [1] -
Machiguchi, T.; Okamoto, J.; Morita, Y.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2006, 128, 44–45. doi:10.1021/ja054619i
Return to citation in text: [1] [2] -
Machiguchi, T.; Okamoto, J.; Takachi, J.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2003, 125, 14446–14448. doi:10.1021/ja030191g
Return to citation in text: [1] [2] -
England, D. C.; Krespan, C. G. J. Am. Chem. Soc. 1965, 87, 4019–4020. doi:10.1021/ja01095a066
Return to citation in text: [1] -
England, D. C.; Krespan, C. G. J. Org. Chem. 1970, 35, 3312–3322. doi:10.1021/jo00835a029
Return to citation in text: [1] -
Scheeren, H. W.; Aben, R. W. M.; Ooms, P. H. J.; Nivard, R. J. F. J. Org. Chem. 1977, 42, 3128–3132. doi:10.1021/jo00439a005
Return to citation in text: [1] -
Dorwald, F. C. Side Reactions in Organic Synthesis; Wiley VCH: Weinheim, 2005; p 144.
Return to citation in text: [1] -
Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals; Elsevier Butterworth Heinemann: Boston, 2003; p 284.
Return to citation in text: [1] [2] [3] -
Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/S0022-1139(01)00375-X
Return to citation in text: [1]
| 9. | Machiguchi, T.; Okamoto, J.; Morita, Y.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2006, 128, 44–45. doi:10.1021/ja054619i |
| 10. | Machiguchi, T.; Okamoto, J.; Takachi, J.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2003, 125, 14446–14448. doi:10.1021/ja030191g |
| 16. | Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/S0022-1139(01)00375-X |
| 15. | Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals; Elsevier Butterworth Heinemann: Boston, 2003; p 284. |
| 1. | Davis, H. R. (Minnesota Mining and Manufacturing Co.). U.S. Patent 3164610, 1965. |
| 2. | Gambaryan, N. P.; Simonyan, L. A.; Petrovskii, P. V. Izv. Akad. Nauk SSSR, Ser. Khim. 1967, 918–921. |
| 3. | Braun, R. A. (du Pont de Nemours, E. I., and Co.). U.S. Patent 3417102, 1968. |
| 14. | Dorwald, F. C. Side Reactions in Organic Synthesis; Wiley VCH: Weinheim, 2005; p 144. |
| 15. | Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals; Elsevier Butterworth Heinemann: Boston, 2003; p 284. |
| 2. | Gambaryan, N. P.; Simonyan, L. A.; Petrovskii, P. V. Izv. Akad. Nauk SSSR, Ser. Khim. 1967, 918–921. |
| 15. | Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals; Elsevier Butterworth Heinemann: Boston, 2003; p 284. |
| 1. | Davis, H. R. (Minnesota Mining and Manufacturing Co.). U.S. Patent 3164610, 1965. |
| 6. | Davis, H. R. (Minnesota Mining and Manufacturing Co.). U.S. Patent 3284509, 1966. |
| 11. | England, D. C.; Krespan, C. G. J. Am. Chem. Soc. 1965, 87, 4019–4020. doi:10.1021/ja01095a066 |
| 12. | England, D. C.; Krespan, C. G. J. Org. Chem. 1970, 35, 3312–3322. doi:10.1021/jo00835a029 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 5. | Middleton, W. J. J. Org. Chem. 1965, 30, 1395–1398. doi:10.1021/jo01016a011 |
| 13. | Scheeren, H. W.; Aben, R. W. M.; Ooms, P. H. J.; Nivard, R. J. F. J. Org. Chem. 1977, 42, 3128–3132. doi:10.1021/jo00439a005 |
| 7. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2009, 130, 780–787. doi:10.1016/j.jfluchem.2009.06.011 |
| 8. | Turro, N. J.; Williams, J. R. Tetrahedron Lett. 1969, 321–323. doi:10.1016/S0040-4039(01)87682-6 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 7. | Petrov, V. A.; Marshall, W. J. Fluorine Chem. 2009, 130, 780–787. doi:10.1016/j.jfluchem.2009.06.011 |
| 9. | Machiguchi, T.; Okamoto, J.; Morita, Y.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2006, 128, 44–45. doi:10.1021/ja054619i |
| 10. | Machiguchi, T.; Okamoto, J.; Takachi, J.; Hasegawa, T.; Yamabe, S.; Minato, T. J. Am. Chem. Soc. 2003, 125, 14446–14448. doi:10.1021/ja030191g |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
| 2. | Gambaryan, N. P.; Simonyan, L. A.; Petrovskii, P. V. Izv. Akad. Nauk SSSR, Ser. Khim. 1967, 918–921. |
| 4. | Kitazume, T.; Otaka, T.; Takei, R.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1976, 49, 2491–2494. doi:10.1246/bcsj.49.2491 |
© 2010 Petrov and Marshall; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)