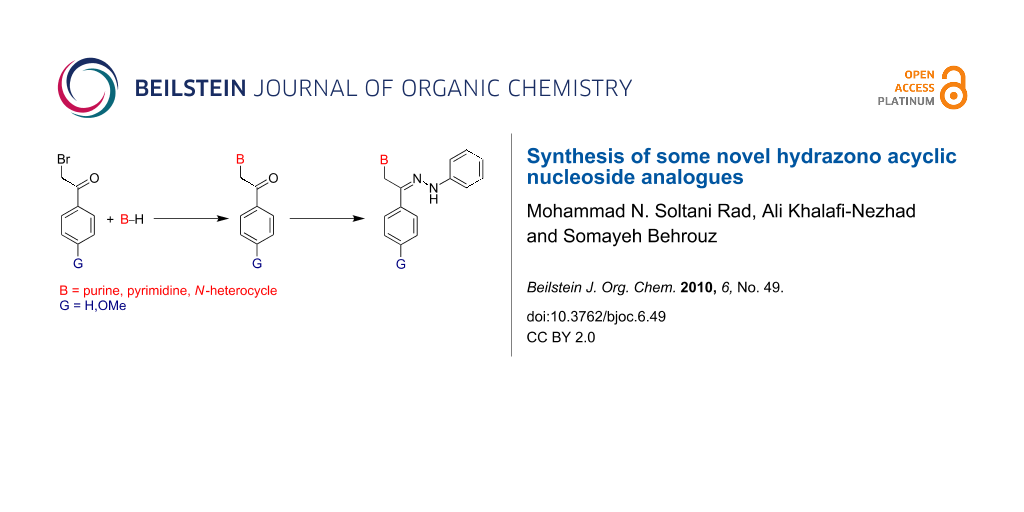
Abstract
The syntheses of novel hydrazono acyclic nucleosides similar to miconazole scaffolds are described. In this series of acyclic nucleosides, pyrimidine as well as purine and other azole derivatives replaced the imidazole function in miconazole and the ether group was replaced with a hydrazone moiety using phenylhydrazine. To interpret the dominant formation of (E)-hydrazone derivatives rather than (Z)-isomers, PM3 semiempirical quantum mechanic calculations were carried out which indicated that the (E)-isomers had the lower heats of formation.

Graphical Abstract
Introduction
In recent years, fungal infections have become an important complication and a major cause of morbidity and mortality in immune-compromised individuals who suffer from tuberculosis, cancer or AIDS, and who undergo organ transplants [1,2]. Up to now, there has been an increase in the number of antifungal drugs available with various structures and scaffolds. However, their clinical value has been limited by the emergence of drug resistance, high risk of toxicity, insufficiencies in their antifungal activity as well as undesirable side effects. This situation has led to an ongoing search for safe and efficient chemotherapeutic agents with potent broad-spectrum antifungal activities.
Some of the best known antifungal azole drugs having rational versatility in structures are miconazole (A), oxiconazole (B) and related compounds, namely aryl azoles (Figure 1).
![[1860-5397-6-49-1]](/bjoc/content/figures/1860-5397-6-49-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of miconazole (A), oxiconazole (B) and hydrazono acyclic nucleoside analogue (C).
Figure 1: Chemical structures of miconazole (A), oxiconazole (B) and hydrazono acyclic nucleoside analogue (C...
Pharmacokinetic properties and cellular permeability of a drug can be modulated by derivatization to bio-reversible forms of the drug, namely hydrazones [3]. Hydrazone derivatives are prominent structural motifs in numerous pharmaceutically active compounds. Many well known drugs with various clinical activities, such as chemotherapeutic (e.g. nitrofurantoin [4,5]), antihypertensive (e.g. endralazine [4,6]), antibiotic (e.g. rifampicin [4]), tuberculostatic (e.g. glyconiazide [4,7,8]), skeletal muscle relaxant and antispasmodic (e.g. dantrolene [4,9]), antiseptic (e.g. nitrofural [4]) and intercalating antineoplastic (e.g. bisantrene [4,10]), contain a hydrazone moiety in their structure. Furthermore, various structurally related miconazole bioactive hydrazones are known as antimicrobial and antifungal agents [11,12].
The significance of nucleoside chemistry in drug discovery is well-known and fully established in medicinal chemistry [13-16]. Acyclic nucleosides constitute a special class of nucleoside analogues which have attracted great interest due to their broad-spectrum chemotherapeutic activities against cancer and infections caused by viruses, microbes and other pathogenic microorganisms. Moreover, antitumor and antiviral activities of various hydrazone derivatives of nucleosides have also been reported [16-18].
Inspired by the miconazole scaffold and also as an extension of our ongoing research in the design and synthesis of novel acyclic nucleosides [19-25], hereby, we wish to report the synthesis of some novel hydrazono acyclic nucleosides having similar scaffolds to the miconazole framework. In these compounds, the nucleobases including pyrimidines, purines and other azole derivatives were substituted as heterocyclic cores and the ether bond in miconazole (i.e. CHOCH2) was replaced with hydrazono bond (i.e. C=NNH) in the newly synthesized compounds. Figure 1 shows the general structure of hydrazono acyclic nucleoside compound C.
Results and Discussion
The synthetic pathways for compounds 2a–2g and 2h–2o are outlined in Scheme 1 and Scheme 2. Two different strategies were considered for the synthesis of the title compounds taking into account the differences in chemical behavior of purine and pyrimidine nucleobases compared to azoles. Because of their better solubility, reactivity and ease of separation of products, the reactions of the azole derivatives were conducted according to Scheme 1.
![[1860-5397-6-49-i1]](/bjoc/content/inline/1860-5397-6-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathway for hydrazono acyclic nucleoside 2a–2g.
Scheme 1: Synthetic pathway for hydrazono acyclic nucleoside 2a–2g.
As shown in Scheme 1, condensation of imidazole, 2-phenylimidazole, hydantoin, benzimidazole and benzotriazole with 2-bromoacetophenone and 4′-methoxy-2-bromoacetophenone gave the key intermediates, ketones 1a–1g. Based on our literature survey, we recognized that among published methods for N-alkylation of imidazole derivatives [26-34], the method developed by Liu et al. [35] was the most appropriate one for N-alkylation of azoles and their derivatives, since in this method the formation of quaternary imidazolium salts is largely prevented. However, from our experience, using triethylamine (TEA) as a homogeneous base instead of K2CO3 (which was previously employed by Liu et al.) produced more satisfactory results. Hence, the reaction of imidazole or benzimidazole derivatives with 2-bromoacetophenones and TEA in the presence of a catalytic amount of n-tetrabutylammonium bromide (TBAB) in refluxing anhydrous acetonitrile (MeCN) provided ketones 1a–1g in good yields (60–70%). Subsequently, the ketones 1a–1g were converted to the pure hydrazone derivatives 2a–2g by treatment with phenylhydrazine in the presence of a catalytic amount of acetic acid in ethanol, followed by heating at reflux (Scheme 1).
For the synthesis of the purine and pyrimidine analogues of target compounds 2h–2o, we envisaged that there might be substantial problems in the synthesis of the desired ketones using similar method as outlined in Scheme 1. This proved to be the case, and the corresponding ketones 1h–1o could not be obtained satisfactorily by this method. The main limitation of using the aforementioned pathway for purines and pyrimidines is their low solubility in acetonitrile, which results in low yields of the corresponding ketones 1h–1o. Thus, we have modified Scheme 1 by using DMF as the solvent of choice for nucleobases (Scheme 2).
![[1860-5397-6-49-i2]](/bjoc/content/inline/1860-5397-6-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic pathway for hydrazono acyclic nucleoside 2h–2o.
Scheme 2: Synthetic pathway for hydrazono acyclic nucleoside 2h–2o.
Thus, the condensation of purine and pyrimidine nucleobases as well as theophylline and theobromine with 2-bromoacetophenones using K2CO3 in anhydrous DMF under reflux conditions provides the N-alkylation adducts 1h–1o in moderate yields (Scheme 2). Subsequently, treatment of the obtained ketones 1h–1o with phenylhydrazine in refluxing ethanol in the presence of a catalytic amount of acetic acid afforded the desired compounds 2h–2o. The structures of synthesized compounds are shown in Table 1. As Table 1 indicates, the N-alkylation reactions of nucleobases were achieved regioselectively. In the case of pyrimidine nucleobases, N(1)-alkylated compounds 1h–1l were the major products (43%, 40%, 51%, 47% and 28%); however, N(1),N(3)-dialkylated adduct was also observed in trace amounts (<10%). Moreover, N-benzyl adenine derivative 1o was obtained as the N(9)-isomer in 76% yield, while theophylline was mostly alkylated at N(7) to afford 1m (69%).
Table 1: Synthesized hydrazono acyclic nucleoside analogues.
| Entry | Product (2) | mp (°C) | Yielda (%) |
|---|---|---|---|
| a |
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-49-i3.svg?max-width=637&scale=1.0)
|
169.2 | 87 |
| b |
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-49-i4.svg?max-width=637&scale=1.0)
|
171.3 | 84 |
| c |
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-49-i5.svg?max-width=637&scale=1.0)
|
174.5 | 82 |
| d |
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-49-i6.svg?max-width=637&scale=1.0)
|
200.1 | 89 |
| e |
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-49-i7.svg?max-width=637&scale=1.0)
|
165.9 | 75 |
| f |
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-49-i8.svg?max-width=637&scale=1.0)
|
148.4 | 81 |
| g |
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-49-i9.svg?max-width=637&scale=1.0)
|
137.9 | 82 |
| h |
![[Graphic 8]](/bjoc/content/inline/1860-5397-6-49-i10.svg?max-width=637&scale=1.0)
|
300.1 | 78 |
| i |
![[Graphic 9]](/bjoc/content/inline/1860-5397-6-49-i11.svg?max-width=637&scale=1.0)
|
213.8 | 80 |
| j |
![[Graphic 10]](/bjoc/content/inline/1860-5397-6-49-i12.svg?max-width=637&scale=1.0)
|
205.1 | 75 |
| k |
![[Graphic 11]](/bjoc/content/inline/1860-5397-6-49-i13.svg?max-width=637&scale=1.0)
|
181.5 | 79 |
| l |
![[Graphic 12]](/bjoc/content/inline/1860-5397-6-49-i14.svg?max-width=637&scale=1.0)
|
173.9 | 74 |
| m |
![[Graphic 13]](/bjoc/content/inline/1860-5397-6-49-i15.svg?max-width=637&scale=1.0)
|
100.1 | 81 |
| n |
![[Graphic 14]](/bjoc/content/inline/1860-5397-6-49-i16.svg?max-width=637&scale=1.0)
|
104.7 | 71 |
| o |
![[Graphic 15]](/bjoc/content/inline/1860-5397-6-49-i17.svg?max-width=637&scale=1.0)
|
209.3 | 84 |
aIsolated yield.
All compounds were fully characterized and their structures confirmed by 1H and 13C NMR, elemental analysis, mass and IR spectroscopy. Although compounds 2a–2o are expected to be produced as two geometrical isomers ((E)- or (Z)-isomer), 1H and 13C NMR analysis indicated that the (E)-isomer was obtained as major product; the minor (Z)-isomer was detected in trace amounts (<5%). To interpret the preference for the (E)-isomers, PM3 semiempirical quantum mechanical calculations were carried out using MOPAC in CS Chem 3D Ultra 8 (Cambridge Soft, 2004) or Hyperchem (Hypercube Inc., Version 7). The results are summarized in Table 2; ΔE refers to the energy difference between the (Z)- and (E)-isomer (ΔE = EZ − EE (kcal/mol)). As can be seen in Table 2, calculated ΔE values for all hydrazone derivatives 2a–2o are positive. There is accord between the experimental observations and calculated data (Table 2) which supports the greater stability of the (E)-isomers over the (Z)-isomers and hence predominant formation of the (E)-products.
Table 2: Calculated heat of formation of synthesized hydrazones 2a–2o using PM3.
| Entry | EEa | EZb | ΔEc |
|---|---|---|---|
| 2a | 123.87244 | 129.56016 | 5.68772 |
| 2b | 85.14540 | 91.62108 | 6.47568 |
| 2c | 148.47301 | 153.04860 | 4.57559 |
| 2d | 14.42535 | 21.51980 | 7.09445 |
| 2e | 142.60529 | 146.16960 | 3.56431 |
| 2f | 103.74787 | 117.44982 | 13.70195 |
| 2g | 183.57186 | 198.25266 | 14.68080 |
| 2h | 26.07071 | 28.66346 | 2.59275 |
| 2i | −13.34542 | −9.56043 | 3.78499 |
| 2j | 21.58339 | 24.58239 | 2.99900 |
| 2k | 17.75077 | 20.31085 | 2.56008 |
| 2l | 52.80499 | 55.83149 | 3.02560 |
| 2m | 21.96581 | 45.90909 | 23.94328 |
| 2n | 52.57276 | 60.37796 | 7.80520 |
| 2o | 176.15539 | 185.18056 | 9.02517 |
aHeat of formation of (E)-isomer (kcal/mol).
bHeat of formation of (Z)-isomer (kcal/mol).
cΔE = EZ − EE (kcal/mol).
Conclusion
In summary, we have synthesized novel acyclic nucleosides 2a–2o containing a miconazole-like scaffold as well as hydrazone moiety. In this series of acyclic nucleosides, we employed various nucleobases and other imidazole derivatives in place of the imidazole in miconazole. To interpret the dominant formation of the (E)-hydrazone derivatives rather than the (Z)-isomers, PM3 semiempirical quantum mechanical calculations were carried out which showed that the heats of formation of the (E)-isomers were lower.
The biological studies of compounds 2a–2o are currently under investigation and will be reported in due course.
Experimental
General
All chemicals were purchased from Fluka or Merck. Solvents were purified and dried according to the reported methods [36] and stored over 0.3 nm molecular sieves. The progress of reaction was followed by TLC using silica gel SILG/UV 254 plates. Silica gel 60, 0.063–0.200 mm (70–230 mesh ASTM) was used for column chromatography. IR spectra were run on a Shimadzu FTIR-8300 spectrophotometer. The 1H NMR (250 MHz) and 13C NMR (62.5 MHz) were run on a Bruker Avance DPX-250, FT-NMR spectrometer (δ in ppm, J in Hz). Mass spectra were recorded on a Shimadzu GC MS-QP 1000 EX apparatus. Microanalyses were performed on a Perkin-Elmer 240-B microanalyzer. Melting points (mp) were recorded on a Büchi 510 apparatus in open capillary tubes and are uncorrected.
General procedure for the synthesis of ketones 1a–1g
In a two-neck round-bottom flask (100 mL) equipped with a condenser, a mixture of the appropriate N-heterocycle (0.01 mol), the 2-bromoacetophenone (0.012 mol), anhydrous TEA (1.01 g, 0.01 mol) and a catalytic amount of TBAB (0.1 g) was dissolved in dry acetonitrile (40 mL). The mixture was then heated at reflux for 10 h (TLC control). The solvent was evaporated at reduced pressure, the residue dissolved in CHCl3 (200 mL) and washed with H2O (2 × 100 mL). The organic layer was dried (10 g of Na2SO4) and concentrated to afford the crude product, which was purified by column chromatography on silica gel eluting with an appropriate solvent.
General procedure for the synthesis of ketones 1h–1o
In a two-neck round-bottom flask (100 mL) equipped with a condenser, a mixture of the appropriate nucleobase (0.01 mol), the 2-bromoacetophenone (0.012 mol), K2CO3 (1.38 g, 0.01 mol) and a catalytic amount of TBAB (0.1 g) was dissolved in dry DMF (30 mL). The mixture was then heated at reflux for 2–3 h (TLC control). The solvent was evaporated at reduced pressure, the residue dissolved in CHCl3 (200 mL) and washed with H2O (2 × 100 mL). The organic layer was dried (10 g of Na2SO4) and concentrated to afford the crude product, which was purified by column chromatography on silica gel eluting with an appropriate solvent.
General procedure for the synthesis of hydrazono acyclic nucleoside analogues 2a–2o
In a two-neck round-bottom flask (100 mL) equipped with a condenser, a mixture of the appropriate ketone 1a–1o (0.01 mol), phenylhydrazine (1.62 g, 0.015 mol) and acetic acid (3 drops) was dissolved in ethanol (15 mL) and the mixture heated at reflux for 15 h (TLC control). The reaction mixture was then stored in a refrigerator overnight. It was filtered and washed with cold ethanol (2 × 5 mL) and recrystallized from MeOH/H2O to afford the pure phenylhydrazone derivatives.
Supporting Information
Supporting information features physical and spectroscopic data for all novel compounds 2a–2o.
| Supporting Information File 1: Synthesis of some novel hydrazono acyclic nucleoside analogues | ||
| Format: PDF | Size: 75.3 KB | Download |
References
-
Georgopapadakou, N. H.; Walsh, T. J. Antimicrob. Agents Chemother. 1996, 40, 279–291.
Return to citation in text: [1] -
Fisher-Hoch, S. P.; Hutwagner, L. Clin. Infect. Dis. 1995, 21, 897–904.
Return to citation in text: [1] -
Maccari, R.; Ottanà, R.; Monforte, F.; Vigorita, M. G. Antimicrob. Agents Chemother. 2002, 46, 294–299. doi:10.1128/AAC.46.2.294-299.2002
Return to citation in text: [1] -
Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Szarvasi, E.; Fontaine, L.; Betbeder-Matibet, A. J. Med. Chem. 1973, 16, 281–287. doi:10.1021/jm00261a027
Return to citation in text: [1] -
Schenker, E.; Salzmann, R. Arzneim. Forsch. 1979, 29, 1835–1843.
Return to citation in text: [1] -
Pennington, F. C.; Guercio, P. A.; Solomons, I. A. J. Am. Chem. Soc. 1953, 75, 2261. doi:10.1021/ja01105a514
Return to citation in text: [1] -
Sah, P. P. T. J. Am. Chem. Soc. 1953, 75, 2512–2513. doi:10.1021/ja01106a516
Return to citation in text: [1] -
Snyder, H. R., Jr.; Davis, C. S.; Bickerton, R. K.; Halliday, R. P. J. Med. Chem. 1967, 10, 807–810. doi:10.1021/jm00317a011
Return to citation in text: [1] -
Murdock, K. C.; Child, R. G.; Lin, Y. I.; Warren, J. D.; Fabio, P. F.; Lee, V. J.; Izzo, P. T.; Lang, S. A., Jr.; Angier, R. B.; Citarella, R. V.; Wallace, R. E.; Durr, F. E. J. Med. Chem. 1982, 25, 505–518. doi:10.1021/jm00347a006
Return to citation in text: [1] -
Mamolo, M. G.; Zampieri, D.; Falagiani, V.; Vio, L.; Fermeglia, M.; Ferrone, M.; Pricl, S.; Banfi, E.; Scialinoc, G. ARKIVOC 2004, v, 231–250.
Return to citation in text: [1] -
Dyer, R. L.; Ellames, G. J.; Hamill, B. J.; Manley, P. W.; Pope, A. M. S. J. Med. Chem. 1983, 26, 442–445. doi:10.1021/jm00357a023
Return to citation in text: [1] -
De Clercq, E. In Advances in Antiviral Drug Design; Johnsson, N. G., Ed.; JAI Press: Greenwich, 1993; Vol. 1, p 88.
Return to citation in text: [1] -
Pathak, T. Chem. Rev. 2002, 102, 1623–1668. doi:10.1021/cr0104532
Return to citation in text: [1] -
Agrofoglio, L. A.; Gillaizeau, I.; Saito, Y. Chem. Rev. 2003, 103, 1875–1916. doi:10.1021/cr010374q
Return to citation in text: [1] -
Huryn, D. M.; Okabe, M. Chem. Rev. 1992, 92, 1745–1768. doi:10.1021/cr00016a004
Return to citation in text: [1] [2] -
De Clercq, E. Nucleosides Nucleotides 1994, 13, 1271–1295. doi:10.1080/15257779408012151
Return to citation in text: [1] -
Magdalena, J.; Fernández, S.; Ferrero, M.; Gotor, V. J. Org. Chem. 1998, 63, 8873–8879. doi:10.1021/jo981062z
Return to citation in text: [1] -
Soltani Rad, M. N.; Khalafi-Nezhad, A.; Behrouz, S.; Asrari, Z.; Behrouz, M.; Amini, Z. Synthesis 2009, 3067–3076. doi:10.1055/s-0029-1216887
Return to citation in text: [1] -
Soltani Rad, M. N.; Khalafi-Nezhad, A.; Behrouz, S.; Faghihi, M. A.; Zare, A.; Parhami, A. Tetrahedron 2008, 64, 1778–1785. doi:10.1016/j.tet.2007.11.101
Return to citation in text: [1] -
Khalafi-Nezhad, A.; Soltani Rad, M. N.; Moosavi-Movahedi, A. A.; Kosari, M. Helv. Chim. Acta 2007, 90, 730–737. doi:10.1002/hlca.200790073
Return to citation in text: [1] -
Khalafi-Nezhad, A.; Soltani Rad, M. N.; Khoshnood, A. Synthesis 2004, 583–589. doi:10.1055/s-2004-815968
Return to citation in text: [1] -
Khalafi-Nezhad, A.; Zarea, A.; Soltani Rad, M. N.; Mokhtari, B.; Parhami, A. Synthesis 2005, 419–424. doi:10.1055/s-2004-834950
Return to citation in text: [1] -
Khalafi-Nezhad, A.; Soltani Rad, M. N.; Hakimelahi, G. H.; Mokhtari, B. Tetrahedron 2002, 58, 10341–10344. doi:10.1016/S0040-4020(02)01415-1
Return to citation in text: [1] -
Khalafi-Nezhad, A.; Zare, A.; Parhami, A.; Soltani Rad, M. N. ARKIVOC 2006, xii, 161–172.
Return to citation in text: [1] -
Nardi, D.; Tajana, A.; Leonardi, A.; Pennini, R.; Portioli, F.; Magistretti, M. J.; Subissi, A. J. Med. Chem. 1981, 24, 727–731. doi:10.1021/jm00138a017
Return to citation in text: [1] -
Hofmann, K. The Chemistry of Heterocyclic Compounds: Imidazole and its Derivatives; Interescience: London, 1953; Part 1.
Return to citation in text: [1] -
Grimmett, M. R. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Pergamon: Oxford, 1996; Vol. 3, p 77.
Return to citation in text: [1] -
Grimmett, M. R. Adv. Heterocycl. Chem. 1970, 12, 103–183. doi:10.1016/S0065-2725(08)60973-3
Return to citation in text: [1] -
Hodges, R.; Grimmett, M. R. Aust. J. Chem. 1968, 21, 1085–1087.
Return to citation in text: [1] -
Häring, M. Helv. Chim. Acta 1959, 42, 1845–1846. doi:10.1002/hlca.19590420612
Return to citation in text: [1] -
Mathias, L. J.; Burkett, D. Tetrahedron Lett. 1979, 20, 4709–4712. doi:10.1016/S0040-4039(01)86690-9
Return to citation in text: [1] -
Kikugawa, Y. Synthesis 1981, 124–125.
Return to citation in text: [1] -
de Savignac, A.; Roques, C.; Hinedi, M.; Michel, G.; Lattes, A. Eur. J. Med. Chem. 1990, 25, 449–454. doi:10.1016/0223-5234(90)90009-R
Return to citation in text: [1] -
Liu, Z.-Z.; Chen, H.-C.; Cao, S.-L.; Li, R.-T. Synth. Commun. 1993, 23, 2611–2615. doi:10.1080/00397919308012596
Return to citation in text: [1] -
Vogel, A. I. Practical Organic Chemistry; Longmans, Green and Co.: London, England, 1954; Chapter 2, pp 161–176.
Return to citation in text: [1]
| 1. | Georgopapadakou, N. H.; Walsh, T. J. Antimicrob. Agents Chemother. 1996, 40, 279–291. |
| 2. | Fisher-Hoch, S. P.; Hutwagner, L. Clin. Infect. Dis. 1995, 21, 897–904. |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 35. | Liu, Z.-Z.; Chen, H.-C.; Cao, S.-L.; Li, R.-T. Synth. Commun. 1993, 23, 2611–2615. doi:10.1080/00397919308012596 |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 6. | Schenker, E.; Salzmann, R. Arzneim. Forsch. 1979, 29, 1835–1843. |
| 36. | Vogel, A. I. Practical Organic Chemistry; Longmans, Green and Co.: London, England, 1954; Chapter 2, pp 161–176. |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 5. | Szarvasi, E.; Fontaine, L.; Betbeder-Matibet, A. J. Med. Chem. 1973, 16, 281–287. doi:10.1021/jm00261a027 |
| 19. | Soltani Rad, M. N.; Khalafi-Nezhad, A.; Behrouz, S.; Asrari, Z.; Behrouz, M.; Amini, Z. Synthesis 2009, 3067–3076. doi:10.1055/s-0029-1216887 |
| 20. | Soltani Rad, M. N.; Khalafi-Nezhad, A.; Behrouz, S.; Faghihi, M. A.; Zare, A.; Parhami, A. Tetrahedron 2008, 64, 1778–1785. doi:10.1016/j.tet.2007.11.101 |
| 21. | Khalafi-Nezhad, A.; Soltani Rad, M. N.; Moosavi-Movahedi, A. A.; Kosari, M. Helv. Chim. Acta 2007, 90, 730–737. doi:10.1002/hlca.200790073 |
| 22. | Khalafi-Nezhad, A.; Soltani Rad, M. N.; Khoshnood, A. Synthesis 2004, 583–589. doi:10.1055/s-2004-815968 |
| 23. | Khalafi-Nezhad, A.; Zarea, A.; Soltani Rad, M. N.; Mokhtari, B.; Parhami, A. Synthesis 2005, 419–424. doi:10.1055/s-2004-834950 |
| 24. | Khalafi-Nezhad, A.; Soltani Rad, M. N.; Hakimelahi, G. H.; Mokhtari, B. Tetrahedron 2002, 58, 10341–10344. doi:10.1016/S0040-4020(02)01415-1 |
| 25. | Khalafi-Nezhad, A.; Zare, A.; Parhami, A.; Soltani Rad, M. N. ARKIVOC 2006, xii, 161–172. |
| 3. | Maccari, R.; Ottanà, R.; Monforte, F.; Vigorita, M. G. Antimicrob. Agents Chemother. 2002, 46, 294–299. doi:10.1128/AAC.46.2.294-299.2002 |
| 26. | Nardi, D.; Tajana, A.; Leonardi, A.; Pennini, R.; Portioli, F.; Magistretti, M. J.; Subissi, A. J. Med. Chem. 1981, 24, 727–731. doi:10.1021/jm00138a017 |
| 27. | Hofmann, K. The Chemistry of Heterocyclic Compounds: Imidazole and its Derivatives; Interescience: London, 1953; Part 1. |
| 28. | Grimmett, M. R. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Pergamon: Oxford, 1996; Vol. 3, p 77. |
| 29. | Grimmett, M. R. Adv. Heterocycl. Chem. 1970, 12, 103–183. doi:10.1016/S0065-2725(08)60973-3 |
| 30. | Hodges, R.; Grimmett, M. R. Aust. J. Chem. 1968, 21, 1085–1087. |
| 31. | Häring, M. Helv. Chim. Acta 1959, 42, 1845–1846. doi:10.1002/hlca.19590420612 |
| 32. | Mathias, L. J.; Burkett, D. Tetrahedron Lett. 1979, 20, 4709–4712. doi:10.1016/S0040-4039(01)86690-9 |
| 33. | Kikugawa, Y. Synthesis 1981, 124–125. |
| 34. | de Savignac, A.; Roques, C.; Hinedi, M.; Michel, G.; Lattes, A. Eur. J. Med. Chem. 1990, 25, 449–454. doi:10.1016/0223-5234(90)90009-R |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 10. | Murdock, K. C.; Child, R. G.; Lin, Y. I.; Warren, J. D.; Fabio, P. F.; Lee, V. J.; Izzo, P. T.; Lang, S. A., Jr.; Angier, R. B.; Citarella, R. V.; Wallace, R. E.; Durr, F. E. J. Med. Chem. 1982, 25, 505–518. doi:10.1021/jm00347a006 |
| 13. | De Clercq, E. In Advances in Antiviral Drug Design; Johnsson, N. G., Ed.; JAI Press: Greenwich, 1993; Vol. 1, p 88. |
| 14. | Pathak, T. Chem. Rev. 2002, 102, 1623–1668. doi:10.1021/cr0104532 |
| 15. | Agrofoglio, L. A.; Gillaizeau, I.; Saito, Y. Chem. Rev. 2003, 103, 1875–1916. doi:10.1021/cr010374q |
| 16. | Huryn, D. M.; Okabe, M. Chem. Rev. 1992, 92, 1745–1768. doi:10.1021/cr00016a004 |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 16. | Huryn, D. M.; Okabe, M. Chem. Rev. 1992, 92, 1745–1768. doi:10.1021/cr00016a004 |
| 17. | De Clercq, E. Nucleosides Nucleotides 1994, 13, 1271–1295. doi:10.1080/15257779408012151 |
| 18. | Magdalena, J.; Fernández, S.; Ferrero, M.; Gotor, V. J. Org. Chem. 1998, 63, 8873–8879. doi:10.1021/jo981062z |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 9. | Snyder, H. R., Jr.; Davis, C. S.; Bickerton, R. K.; Halliday, R. P. J. Med. Chem. 1967, 10, 807–810. doi:10.1021/jm00317a011 |
| 4. | Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 3rd ed.; Thieme: Stuttgart, 1999. |
| 7. | Pennington, F. C.; Guercio, P. A.; Solomons, I. A. J. Am. Chem. Soc. 1953, 75, 2261. doi:10.1021/ja01105a514 |
| 8. | Sah, P. P. T. J. Am. Chem. Soc. 1953, 75, 2512–2513. doi:10.1021/ja01106a516 |
| 11. | Mamolo, M. G.; Zampieri, D.; Falagiani, V.; Vio, L.; Fermeglia, M.; Ferrone, M.; Pricl, S.; Banfi, E.; Scialinoc, G. ARKIVOC 2004, v, 231–250. |
| 12. | Dyer, R. L.; Ellames, G. J.; Hamill, B. J.; Manley, P. W.; Pope, A. M. S. J. Med. Chem. 1983, 26, 442–445. doi:10.1021/jm00357a023 |
© 2010 Soltani Rad et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)