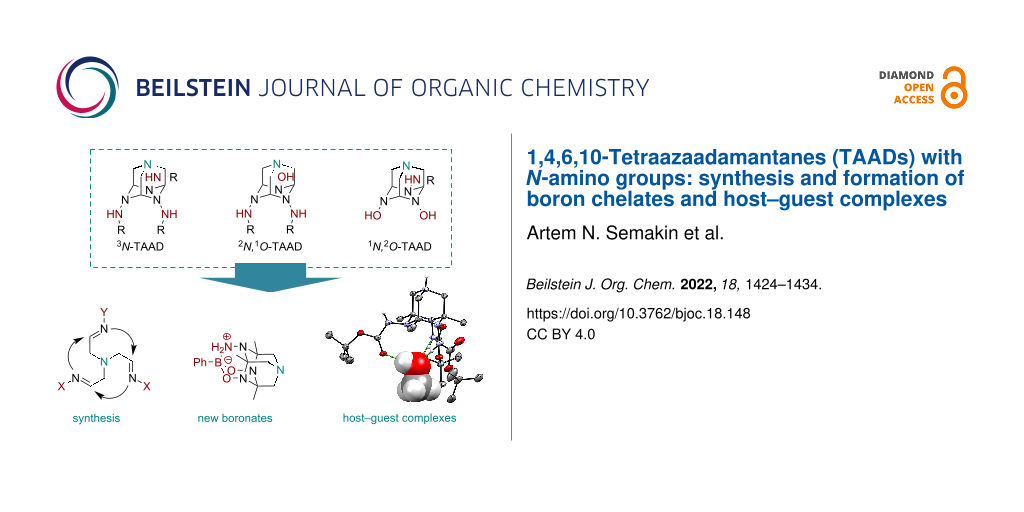
Abstract
A synthetic route to 1,4,6,10-tetraazaadamantanes (TAADs) bearing free and protected amino groups at the bridge N-atoms has been developed via intramolecular cyclotrimerization of C=N units in the corresponding tris(hydrazonoalkyl)amines. In a similar fashion, unsymmetrically substituted TAADs having both amino and hydroxy groups at the bridge N-atoms were prepared via a hitherto unknown co-trimerization of oxime and hydrazone groups. The use of N-TAAD derivatives as potential ligands and receptors was showcased through forming boron chelates and host–guest complexes with water and simple alcohols.

Graphical Abstract
Introduction
Tripodal molecules are widely used as chelating ligands for transition metal catalysis [1-3], sensors for ions and small molecules [4,5], reagents for surface grafting [6], building blocks for the construction of supramolecular structures [7], polycyclic cage molecules [8], and porous materials [9]. In tripodal molecules, three functional arms are bound to a central trivalent scaffold, which can be a single atom (e.g., carbon, nitrogen, or phosphorus), a small ring (usually aromatic or saturated six-membered ring), or a macrocycle (Figure 1a). The functional properties of tripodal molecules are governed not only by the nature and length of the side arms but also by the size and geometry of the central scaffold [2,4]. By changing the scaffold, the volume of the cavity created by the three arms can be tuned allowing for the coordination of ions and molecules of different sizes [4]. For this reason, the central scaffold should also have a rigid geometry without much conformational flexibility to create a preorganized binding pocket. In this regard, the adamantane scaffold received some attention as it represents a conformationally strained analog of the cyclohexane ring [10-13]. However, the preparation of trisubstituted adamantanes is challenging because multiple functionalization of the parent hydrocarbon is not selective, while the synthesis from acyclic precursors requires multistep synthetic routes [10,14]. The use of azaadamantanes is more advantageous as they are prepared via self-assembly reactions from simple precursors and easily functionalized through addition reactions at heteroatoms [15-20].
![[1860-5397-18-148-1]](/bjoc/content/figures/1860-5397-18-148-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Adamantane-based tripodal scaffolds and current work.
Figure 1: Adamantane-based tripodal scaffolds and current work.
Some time ago, our group introduced 1,4,6,10-tetraazaadamantane (TAAD) as a promising molecular platform for the design of functional tripodal molecules (Figure 1b) [21-23]. In particular, N,N,N-trihydroxy-TAAD derivatives (3O-TAADs) were shown to be chelating ligands for boron [24]. The application of TAAD-boronate complexes was demonstrated by the preparation of conjugates of boronic acids with biomolecules [25,26], COF-like materials [25], and dynamic covalent libraries [25]. TAAD can be covalently bound to a polymer matrix through the nucleophilic nitrogen N(1) that was used to prepare scavengers of boronic acids [25]. Also, TAAD was demonstrated to serve as a scorpionate-type ligand for manganese and iron leading to complexes with the metal in an atypical +4 oxidation state [27-29]. Recently, TAAD was used to improve the thermal and photochemical stability of MAPbI3 perovskite films (most likely through coordination of Pb(II) ions) [30]. The disadvantages of 3O-TAADs, however, are limited thermal stability (due to cage opening to corresponding linear tris-oximes) and difficulties in the functionalization of N-hydroxy groups [21].
Currently, our research is focusing on TAAD derivatives bearing protected and free amino groups at the bridge nitrogen atoms (N-TAADs, Figure 1c). Our main interest in these products was due to their potential as synthetic receptors to form host–guest complexes based on H-bond interactions with amide/carbamoyl groups [31,32]. In addition, N-TAADs can be viewed as analogs of hexahydrazide ligands, which were reported to stabilize high-valent metal complexes [33,34]. Since N-amino groups can be easily functionalized through the addition of electrophilic reagents, various functional arms can be attached. Moreover, N-TAAD derivatives were expected to be less prone to cage-opening reaction compared to 3O-TAADs.
Here, we wish to report the synthesis of TAADs decorated with N-amino(amido) groups (3N-TAADs) by cyclization of the corresponding tris-hydrazones, as well as the assembly of unsymmetrically substituted TAADs having both amino(amido) and hydroxy groups at bridge nitrogen atoms (2N,1O-TAADs and 1N,2O-TAADs) via a hitherto unknown co-trimerization of oxime and hydrazone units [35]. Also, structural studies of the obtained TAAD derivatives were performed, and the formation of boron chelates and host–guest complexes having an unusual intramolecular H-bonded network was showcased in this work.
Results and Discussion
Synthesis of 3N-TAADs, 2N,1O-TAADs and 1N,2O-TAADs
Our strategy to construct TAAD derivatives involves the intramolecular cyclotrimerization of C=N bonds in a suitable trisimine precursor (Scheme 1). Previously, 3O-TAADs 2 were prepared by cyclization of corresponding trisoximes 1 [21]. However, this reaction is slow and reversible (upon heating adamantane structure 2 reverts to the tris-oxime form 1). Previous experimental and computational data evidence that cyclotrimerization of hydrazone groups proceeds more readily compared to oximes [18]. Hence, hydrazones 3 were expected to cyclize to corresponding TAADs 4 with high efficiency. On the other hand, cyclization of mixed oxime-hydrazone acyclic precursors 5 and 7 to give 2N,1O-TAADs (6) and 1N,2O-TAADs (8) was ambiguous as no examples of co-trimerization of hydrazone and oxime groups (either inter- or intramolecular) have been reported to the best of our knowledge.
![[1860-5397-18-148-i1]](/bjoc/content/inline/1860-5397-18-148-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A general strategy for the assembly of TAAD derivatives.
Scheme 1: A general strategy for the assembly of TAAD derivatives.
Initially, a series of trishydrazones 3a–e were synthesized by a triple alkylation of ammonia with α-halohydrazones 9a–e following a protocol previously developed by us [35] (Scheme 2a). Halohydrazones 9a–e were prepared by condensation of readily available chloroacetone or bromoacetaldehyde (generated from the corresponding diethyl acetal) with hydrazides or carbazates. In the synthesis of product 3a the intermediate α-chlorohydrazone 9a was not isolated.
![[1860-5397-18-148-i2]](/bjoc/content/inline/1860-5397-18-148-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of acyclic precursors to 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs.
Scheme 2: Synthesis of acyclic precursors to 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs.
The preparation of mixed oxime-hydrazones 5 and 7 was more challenging as it required a modular synthetic approach and the use of protecting groups. The developed synthetic route starting from benzylamine is shown in Scheme 2b and c. To access mixed oxime-hydrazone 5a, benzylamine was reacted with two equivalents of α-chlorohydrazone 9c to give bishydrazone 10, which was debenzylated by hydrogenolysis with Pd–C catalyst. The subsequent introduction of the oximinoalkyl unit was selectively achieved by treatment of secondary amine 11 with ene-nitrosoacetal 12 [36] (Scheme 2b). The synthesis of products 7a,b containing two oxime groups was accomplished via double oximinoalkylation of benzylamine to give dioxime 13 [36], cleavage of the N-benzyl group, and reaction of the secondary amine 14 with α-chlorohydrazones 9c,d (Scheme 2c).
We then studied the cyclization of the obtained trishydrazones 3 and mixed oxime-hydrazones 5 and 7 to the corresponding TAAD derivatives 4, 6, and 8 (Scheme 3). Upon reflux in methanol, tris-hydrazones 3a,c–e underwent smooth intramolecular [2 + 2 + 2]-cyclization to give the corresponding TAADs 4a,c–e in good to high isolated yields. Trishydrazone 3b with X = NHCO2Et was somewhat less reactive, and its conversion to heteroadamantane 4b required more harsh conditions (reflux in water). TAADs 4a and 4c were converted into their corresponding hydrochlorides, which were used for X-ray analysis (vide infra).
![[1860-5397-18-148-i3]](/bjoc/content/inline/1860-5397-18-148-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs. *Yield based on compound 14.
Scheme 3: Synthesis of 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs. *Yield based on compound 14.
Similar to trishydrazones 3, the mixed oxime-hydrazones 5 and 7 cyclized to the corresponding heteroadamantanes 6 and 8 smoothly. Moreover, compound 7b underwent partial cyclization to heteroadamantane 8b already upon preparation. This is surprising as the parent trisoximes 1 cyclized only in the presence of acid as a promoter [21], while with trishydrazones 3 the process takes place only at elevated temperatures. Thus, a synergistic effect of the oxime and hydrazone groups on the intramolecular cyclotrimerization reaction takes place. The structure of 1N,2O-TAAD 8a was secured by X-ray analysis (vide infra).
Unlike 3O-TAAD derivatives 2 [21], the obtained 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs are thermally stable and do not suffer from retro-[2 + 2 + 2]-cyclotrimerization to the open-chain tris-imines upon heating. Thus, the presence of at least one N-amido group stabilizes the azaadamantane form making it more preferable under thermodynamic conditions.
Transformations of 3N-TAADs, 2N,1O-TAADs, and 1N,2O-TAADs
We then focused on the preparation of TAAD derivatives with free amino groups by the removal of the protecting N-acyl or alkoxycarbonyl groups. However, treatment of adamantanes 4a and 4b with excess hydrazine (100–200 equiv) upon heating did not lead to any conversion. Also attempts to deprotect Boc-derivatives 4c, 4e, 6a, and 8a with trifluoroacetic acid or hydrochloric acid (in water or dioxane) were not successful and led to complex mixtures of products. Hydrazinium dihydrochloride was isolated in some cases (confirmed by X-ray, mp, and FT-IR data) demonstrating the degradation of the heteroadamantane cage. Deprotection of Cbz derivatives by hydrogenation over Pd–C was more productive. Thus, hydrogenolysis of product 8b delivered the 1N,2O-TAAD derivative 15 bearing a free amino group (Scheme 4). However, this product was prone to a ring–chain isomerization forming an equilibrating mixture of azaadamantane 15, tris-imine 16, and bicycle 17 in solution as determined by NMR data (see Supporting Information File 1 for details). We were able to shift the equilibrium to the adamantane form by forming a stable boronate adduct 18 having a diamantane structure through the reaction with phenylboronic acid. Note that product 18 contains an unprecedented 2,4-dioxa-1,5,7,10-tetraaza-3-boraadamantane motif (Scheme 4). This result demonstrates that N-amino-substituted TAADs are able to serve as chelating ligands. In recent years, cyclic boron ate-complexes have received much interest as protected forms of boronic acids [37-39].
![[1860-5397-18-148-i4]](/bjoc/content/inline/1860-5397-18-148-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Deprotection of TAAD 8b and subsequent complexation with phenylboronic acid.
Scheme 4: Deprotection of TAAD 8b and subsequent complexation with phenylboronic acid.
From this experiment, it became apparent that TAAD derivatives bearing free N-amino groups are prone to cage opening. To stabilize the tetraazaadamantane cage, quaternization of the tertiary bridge-head nitrogen in Boc-protected TAADs 4c, 4e, 6a, and 8a was performed by benzylation [21,40] (Scheme 5). The obtained N-benzyl salts were quantitatively converted into corresponding deprotected TAADs 19–21 by reflux in water (Scheme 5, blue). Alternatively, removal of Boc groups could be performed by treatment with aqueous HCl to give the corresponding hydrochloride salts, which could be converted into free hydrazines by treatment with excess NaHCO3 in ethanol (as demonstrated for product 19c). Note that TAADs 19–21 with free amino groups are more stable than the corresponding hydrochloride salts, which slowly decompose upon storage at rt. The structures of products 19e·3HCl, 21 and its hydrochloride 21·HCl were confirmed by X-ray diffraction analysis (vide infra). In hydrochloride salts, the primary amino groups were protonated.
![[1860-5397-18-148-i5]](/bjoc/content/inline/1860-5397-18-148-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Quaternization of TAADs 4c, 4e, 6a, and 8a followed by deprotection of N-Boc groups.
Scheme 5: Quaternization of TAADs 4c, 4e, 6a, and 8a followed by deprotection of N-Boc groups.
In most cases, deprotection reactions were performed at low temperatures to prevent degradation of the aminal fragments. Indeed, treatment of Bn-4c with hydrochloric acid at 0 °C resulted in a substitution of one NNH2 unit with the oxygen atom producing product 22, having a hitherto undescribed 4-oxa-1,6,10-triazaadamantane skeleton (Scheme 5, red). Also, hydrazinium dihydrochloride was isolated in this experiment.
Deoxygenation of N–OH units in TAAD Bn-8a was also performed by reflux with Zn in water (Scheme 5, green). The Boc group was removed under these conditions producing TAAD 23 in high yield.
Structure of N-TAADs and the formation of host–guest complexes
Several of the obtained TAAD derivatives were characterized by X-ray analysis (namely, 4a, 4a·HCl, 4b [35], 4c·HCl, Bn-4c, 8a, 19e·3HCl, 21 and 21·HCl, see Figure 2 and Supporting Information File 2). The geometrical parameters of the 1,4,6,10-tetraazaadamantane cage in these products are close to that observed in 3O-TAADs of type 2 [21]. Carbon–carbon bonds fall within a narrow range of 1.52–1.53 Å. Carbon–nitrogen bonds of the triazinane ring are within 1.45–1.50 Å, while the C–N distances involving bridge-head nitrogen N(1) expectedly differ in free amines (1.46–1.47 Å) and quaternary ammonium salts (1.48–1.52 Å).
![[1860-5397-18-148-2]](/bjoc/content/figures/1860-5397-18-148-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: General view of the 1,4,6,10-tetraazaadamantane motif in X-ray structures of the obtained N-TAAD derivatives (representation of non-hydrogen atoms as thermal ellipsoids at 50% probability level). Anions and crystal-solvate molecules are omitted for clarity. (a) 4a·HCl·H2O·MeOH. (b) 4c·HCl·3.5MeOH. (c) Bn-4c·3CD3OD (bromide salt). (d) 8a. (e) 19e·3HCl·H2O. (f) 21·HCl·2H2O.
Figure 2: General view of the 1,4,6,10-tetraazaadamantane motif in X-ray structures of the obtained N-TAAD de...
Remarkably, in all 3N-TAADs (both with protected and free amino groups) two N-substituents are located in axial positions, while the third one is equatorial (Figure 2a–c, and e). Carbon–nitrogen bonds involving the nitrogen atom bearing an equatorial group appear to be somewhat elongated (by ca. 0.02–0.03 Å) compared to other C–N bonds of the triazinane ring. In the 1N,2O-TAADs the amino group is always axial, while OH groups occupy either eq,eq (in 21 and 21·HCl) or ax,eq (in 8a) positions (Figure 2d and f). A similar isomerism is observed in the previously described 3O-TAAD derivatives 2 [21]. In all TAADs, the nitrogen with an equatorial substituent is more deviated (0.55–0.61 Å) from the mean plane created by three carbon atoms of the triazinane ring than the nitrogen atom bearing and axial group (0.37–0.45 Å).
3N-TAADs 4 bearing amide and carbamate groups form inclusion complexes with water or methanol (Figure 3a–d). In the crystal state, a common hydrogen-bonded motif is observed, in which the guest molecule is located in a pocket created by amide/carbamate groups and the triazinane ring of the 3N-TAAD structure that is well-illustrated by Hirshfeld surfaces [41] (Figure 3e and f). The guest molecule (water or methanol) acts as an H-bond acceptor with two axial NH units (distances N···O 2.85–2.93 Å, ![[Graphic 1]](/bjoc/content/inline/1860-5397-18-148-i6.svg?max-width=637&scale=1.18182) NHO 166.4–178.0°) and as an H-bond donor for the oxygen atom of the equatorial amide group (distances O···O 2.69–2.78 Å,
NHO 166.4–178.0°) and as an H-bond donor for the oxygen atom of the equatorial amide group (distances O···O 2.69–2.78 Å, ![[Graphic 2]](/bjoc/content/inline/1860-5397-18-148-i7.svg?max-width=637&scale=1.18182) OHO 163.7–178.1°) forming two
OHO 163.7–178.1°) forming two ![[Graphic 3]](/bjoc/content/inline/1860-5397-18-148-i8.svg?max-width=637&scale=1.18182) -cyclic motifs.
-cyclic motifs.
![[1860-5397-18-148-3]](/bjoc/content/figures/1860-5397-18-148-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) General structure of host–guest complexes of 3N-TAADs with water. (b) The general structure of host–guest complexes of 3N-TAADs with methanol. (c) Host–guest motif in the X-ray structure of 4a·HCl·H2O·MeOH (guest molecule is shown in “spacefill” representation). (d) Host–guest motif in the X-ray structure of 4c·HCl·3.5MeOH (guest molecule is shown in “spacefill” representation). (e) Hirshfeld surface of the TAAD cation in 4a·HCl·H2O·MeOH. (e) Hirshfeld surface of the TAAD cation in 4c·HCl·3.5MeOH.
Figure 3: (a) General structure of host–guest complexes of 3N-TAADs with water. (b) The general structure of ...
There seems to be a preference for the coordination of certain guest molecules depending on the nature of the amide/carbamate groups. Thus, N-propionyl and ethyl carbamate derivatives (4a, 4a·HCl, and 4b) form the aforementioned complexes with water (Figure 3c), although lattice methanol molecules and chloride anions are present in the structure. The larger tris-Boc derivatives 4c·HCl and Bn-4c form the same guest–host complexes with methanol (Figure 3d). Thus, these TAAD-based systems may exhibit molecular recognition properties.
Deprotected N-TAADs such as 19e, 21, and 21·HCl as well as the previously described 3O-TAADs 2 do not form such host–guest complexes, although water and methanol molecules are present in their crystal lattices. Thus, the presence of amide/carbamate groups is essential for the assembly of these supramolecular structures.
The formation of complexes can be evidenced in the solution state by NMR (Figure 4). Thus, 1H NMR spectra of protected tetraazaadamantanes 4 and Bn-4 in DMSO-d6 are rather complicated and contain several sets of broad multiplets. This can be attributed to the presence of several invertomers (inversion of the cage nitrogen atoms) and/or rotamers (restricted rotation around amide C–N bonds), which is well-known for amides and carbamates (Figure 4a). The activation barriers for the rotation across amide C–N bonds in 4a and 4c estimated by DFT (18.4 and 19.4 kcal/mol, respectively, ωB97XD/Def2TZVP, gas phase) are close to those observed experimentally for amides and carbamates [42,43]. Upon changing the solvent to CD3OD or D2O the spectra become much simpler and a picture expected for a Cs symmetrical structure is observed (for example, cf. 1H NMR spectra of Bn-4c in different solvents shown in Figure 4b). Such symmetrization can be explained by a pre-organization of N-amido groups through the coordination of water/alcohol leading to the host–guest complex observed in the X-ray.
![[1860-5397-18-148-4]](/bjoc/content/figures/1860-5397-18-148-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) Dynamic processes in TAADs 4 and complexation with ROH. (b) Fragment of 1H NMR spectra of Bn-4c in DMSO-d6 and CD3OD.
Figure 4: (a) Dynamic processes in TAADs 4 and complexation with ROH. (b) Fragment of 1H NMR spectra of Bn-4c...
The formation of the inclusion complexes H2O@TAADs 4a and MeOH@4c·H+ was additionally explored by DFT calculations at the ωB97XD/Def2TZVP level of theory with basis set superposition error (BSSE) correction (Table 1). Interestingly, in the most energetically favored invertomer ax,ax,ax-4 (Figure 4a) the geometry around the nitrogen atom N(4) is close to planar (![[Graphic 4]](/bjoc/content/inline/1860-5397-18-148-i9.svg?max-width=637&scale=1.18182) CN(4)N 121.1–121.4°,
CN(4)N 121.1–121.4°, ![[Graphic 5]](/bjoc/content/inline/1860-5397-18-148-i10.svg?max-width=637&scale=1.18182) CN(4)C 112.5°). Such flattening is due to the formation of intramolecular bifurcated hydrogen-bonds between the corresponding carbonyl oxygen and NH hydrogens of two axial N-amido groups. The invertomer eq,ax,ax-4a (Figure 4a) lacking this H-bond is less stable by ca. 2 kcal/mol (ΔG°). Upon the formation of inclusion complexes with water/methanol, this H-bond is broken and two new H-bonds are formed resulting in a less strained structure with a normal pyramidal geometry of the N-4 nitrogen atom. The calculated structural metrics of complexes modeled by DFT are close to those observed in the X-ray structures.
CN(4)C 112.5°). Such flattening is due to the formation of intramolecular bifurcated hydrogen-bonds between the corresponding carbonyl oxygen and NH hydrogens of two axial N-amido groups. The invertomer eq,ax,ax-4a (Figure 4a) lacking this H-bond is less stable by ca. 2 kcal/mol (ΔG°). Upon the formation of inclusion complexes with water/methanol, this H-bond is broken and two new H-bonds are formed resulting in a less strained structure with a normal pyramidal geometry of the N-4 nitrogen atom. The calculated structural metrics of complexes modeled by DFT are close to those observed in the X-ray structures.
Table 1: DFT calculated binding energies for the guest–host complexes ROH@4.
| Complex | Calculated binding energy, kcal/mola,b | ||
| ΔE0 | ΔH° | ΔG° | |
| H2O@4a | −14.1 | −12.6 | −2.8 |
| H2O@4c·H+ | −10.8 | −9.1 | +1.8 |
| MeOH@4c·H+ | −12.3 | −10.9 | −0.8 |
| t-BuOH@4c·H+ | −18.0 | −16.4 | −3.7 |
aωB97XD/Def2TZVP with BSSE (gas phase). bRelative energies were calculated as ΔE = ΔE(ROH@ax,ax,ax-4) − E(ax,ax,ax-4) − E(ROH).
The formation of inclusion complexes is thermodynamically favored in the gas phase by 1–4 kcal/mol (ΔG°) showing rather weak bond character in these structures. Indeed, the guest molecules are liberated upon drying of the obtained crystal solvates in a vacuum with gentle heating.
Interestingly, a higher preference in coordination of tert-butanol over water and methanol was observed in competition experiments with TAAD 4c (see Supporting Information File 1 for details). DFT calculations demonstrate that binding energy increases in the series H2O < MeOH < t-BuOH (cf. data in Table 1). This can be explained by additional stabilization of the complex through weak hydrophobic interactions of with tert-butyl groups of Boc. We believe that by adjustment of the substituents on the amide/carbamate groups, receptors for supramolecular sensing of alcohols [44] could be designed based on 3N-TAADs of type 4. Further research in this direction is currently underway.
Conclusion
In conclusion, we developed a synthetic route to 1,4,6,10-tetraazaadamantanes bearing free and protected amino groups at the bridge N-atoms (N-TAADs). It involves the assembly of a tris(iminoalkyl)amine precursor, which undergoes smooth intramolecular cyclotrimerization of the C=N bonds to afford the 1,4,6,10-tetraazaadamantane cage. This route was successfully exploited to prepare unsymmetrically substituted TAADs having both amino(amido) and hydroxy groups at bridge nitrogen atoms via a hitherto unknown co-trimerization of oxime and hydrazone groups. The N-TAADs with acylated amino groups are highly stable. In contrast, unprotected N-TAADs are prone to cage opening and hydrolysis, yet quaternization of the bridge-head nitrogen greatly stabilizes their structure. We have showcased that N-TAADs can serve as platforms for the design of chelating ligands and supramolecular receptors. In particular, we have shown that the 1N,2O-TAAD derivative reacts with phenylboronic acid producing a novel type of boron ate-complex with a rigid diamantane geometry. 3N-TAADs bearing N-acyl and alkoxycarbonyl groups form guest–host complexes with water or methanol, in which the guest molecule is fixed by a system of H-bonds in a pocket created by amide units and the triazinane ring. This creates opportunities for a rational design of supramolecular receptors for alcohols based on 3N-TAAD as a template.
Supporting Information
| Supporting Information File 1: Experimental procedures, NMR spectra, infrared spectra, X-ray data, and computation data. | ||
| Format: PDF | Size: 8.1 MB | Download |
| Supporting Information File 2: Crystallographic information files (CIFs) for compounds 19e·3HCl·H2O, Bn-4c(bromide)·3CD3OD, 21·2D2O, 4a·H2O·0.5MeOH, 4a·HCl·H2O·MeOH, 4c·HCl·3.5MeOH, 8a, 21·HCl·2H2O, and hydrazinium dihydrochloride. CCDC deposition numbers 2190361, 2190362, 2190363, 2182991, 2182992, 2182993, 2182994, 2182995, 2183048. | ||
| Format: ZIP | Size: 3.4 MB | Download |
Acknowledgements
The authors thank Dr. M. E. Minyaev, E. I. Tuzharov, and D. I. Nasyrova from the Department of Structural Studies of N. D. Zelinsky Institute of Organic Chemistry for determination of crystal structures for 4a·H2O·0.5MeOH, 4a·HCl·H2O·MeOH, 4c·HCl·3.5MeOH, 8a, 21·HCl·2H2O, and hydrazinium dihydrochloride.
Funding
This work was supported by the Russian Foundation for Basic Research (grant 20-03-00510_a). X-ray diffraction data for Bn-4c(bromide)·3CD3OD, 19e·3HCl·H2O, and 21·2D2O were collected with financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for molecular composition studies of INEOS RAS.
References
-
Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028
Return to citation in text: [1] -
Hu, X.; Meyer, K. J. Organomet. Chem. 2005, 690, 5474–5484. doi:10.1016/j.jorganchem.2005.07.119
Return to citation in text: [1] [2] -
Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308
Return to citation in text: [1] -
Kuswandi, B.; Nuriman; Verboom, W.; Reinhoudt, D. N. Sensors 2006, 6, 978–1017. doi:10.3390/s6080978
Return to citation in text: [1] [2] [3] -
Wadas, T. J.; Wong, E. H.; Weisman, G. R.; Anderson, C. J. Chem. Rev. 2010, 110, 2858–2902. doi:10.1021/cr900325h
Return to citation in text: [1] -
Valášek, M.; Lindner, M.; Mayor, M. Beilstein J. Nanotechnol. 2016, 7, 374–405. doi:10.3762/bjnano.7.34
Return to citation in text: [1] -
Savyasachi, A. J.; Kotova, O.; Shanmugaraju, S.; Bradberry, S. J.; Ó’Máille, G. M.; Gunnlaugsson, T. Chem 2017, 3, 764–811. doi:10.1016/j.chempr.2017.10.006
Return to citation in text: [1] -
Zhang, G.; Mastalerz, M. Chem. Soc. Rev. 2014, 43, 1934–1947. doi:10.1039/c3cs60358j
Return to citation in text: [1] -
Beuerle, F.; Gole, B. Angew. Chem., Int. Ed. 2018, 57, 4850–4878. doi:10.1002/anie.201710190
Return to citation in text: [1] -
Fleck, C.; Franzmann, E.; Claes, D.; Rickert, A.; Maison, W. Synthesis 2013, 45, 1452–1461. doi:10.1055/s-0033-1338470
Return to citation in text: [1] [2] -
Grillaud, M.; Bianco, A. J. Pept. Sci. 2015, 21, 330–345. doi:10.1002/psc.2719
Return to citation in text: [1] -
Lamanna, G.; Grillaud, M.; Macri, C.; Chaloin, O.; Muller, S.; Bianco, A. Biomaterials 2014, 35, 7553–7561. doi:10.1016/j.biomaterials.2014.05.017
Return to citation in text: [1] -
Fleck, C.; Memmel, E.; Fölsing, M.; Poll, B.; Hackl, T.; Seibel, J.; Maison, W. Eur. J. Org. Chem. 2015, 1696–1710. doi:10.1002/ejoc.201403517
Return to citation in text: [1] -
Ivleva, E. A.; Pogulyaiko, A. V.; Klimochkin, Y. N. Russ. J. Org. Chem. 2018, 54, 1294–1300. doi:10.1134/s107042801809004x
Return to citation in text: [1] -
Balija, A. M.; Kohman, R. E.; Zimmerman, S. C. Angew. Chem., Int. Ed. 2008, 47, 8072–8074. doi:10.1002/anie.200802222
Return to citation in text: [1] -
Kohman, R. E.; Zimmerman, S. C. Chem. Commun. 2009, 794–796. doi:10.1039/b818183g
Return to citation in text: [1] -
Hou, T.; Zhang, J.; Wang, C.; Luo, J. Org. Chem. Front. 2017, 4, 1819–1823. doi:10.1039/c7qo00357a
Return to citation in text: [1] -
Semakin, A. N.; Nelyubina, Y. V.; Ioffe, S. L.; Sukhorukov, A. Y. Eur. J. Org. Chem. 2020, 6723–6735. doi:10.1002/ejoc.202000832
Return to citation in text: [1] [2] -
Suslov, E. V.; Ponomarev, K. Y.; Volcho, K. P.; Salakhutdinov, N. F. Russ. J. Bioorg. Chem. 2021, 47, 1133–1154. doi:10.1134/s1068162021060236
Return to citation in text: [1] -
Dalinger, A. I.; Medved’ko, A. V.; Kalinin, M. A.; Sereda, V. A.; Churakov, A. V.; Vatsadze, S. Z. Russ. Chem. Bull. 2021, 70, 1002–1005. doi:10.1007/s11172-021-3180-9
Return to citation in text: [1] -
Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Semakin, A. N.; Sukhorukov, A. Y.; Nelyubina, Y. V.; Khomutova, Y. A.; Ioffe, S. L.; Tartakovsky, V. A. J. Org. Chem. 2014, 79, 6079–6086. doi:10.1021/jo5007703
Return to citation in text: [1] -
Semakin, A. N.; Sukhorukov, A. Y. Targets Heterocycl. Syst. 2017, 21, 82–97.
Return to citation in text: [1] -
Golovanov, I. S.; Sukhorukov, A. Y.; Nelyubina, Y. V.; Khomutova, Y. A.; Ioffe, S. L.; Tartakovsky, V. A. J. Org. Chem. 2015, 80, 6728–6736. doi:10.1021/acs.joc.5b00892
Return to citation in text: [1] -
Golovanov, I. S.; Mazeina, G. S.; Nelyubina, Y. V.; Novikov, R. A.; Mazur, A. S.; Britvin, S. N.; Tartakovsky, V. A.; Ioffe, S. L.; Sukhorukov, A. Y. J. Org. Chem. 2018, 83, 9756–9773. doi:10.1021/acs.joc.8b01296
Return to citation in text: [1] [2] [3] [4] -
de Vries, R. H.; Viel, J. H.; Kuipers, O. P.; Roelfes, G. Angew. Chem., Int. Ed. 2021, 60, 3946–3950. doi:10.1002/anie.202011460
Return to citation in text: [1] -
Samoľová, E.; Premužić, D.; Plociennik, S.; Hołyńska, M. J. Mol. Struct. 2019, 1176, 366–375. doi:10.1016/j.molstruc.2018.08.100
Return to citation in text: [1] -
Premužić, D.; Hołyńska, M.; Ozarowski, A.; Pietzonka, C.; Roseborough, A.; Stoian, S. A. Inorg. Chem. 2020, 59, 10768–10784. doi:10.1021/acs.inorgchem.0c01242
Return to citation in text: [1] -
Golovanov, I. S.; Leonov, A. V.; Lesnikov, V. K.; Pospelov, E. V.; Frolov, K. V.; Korlyukov, A. A.; Nelyubina, Y. V.; Novikov, V. V.; Sukhorukov, A. Y. Dalton Trans. 2022, 51, 4284–4296. doi:10.1039/d1dt04104e
Return to citation in text: [1] -
Ozerova, V. V.; Zhidkov, I. S.; Boldyreva, A.; Dremova, N. N.; Emelianov, N. A.; Shilov, G. V.; Frolova, L. A.; Kurmaev, E. Z.; Sukhorukov, A. Y.; Aldoshin, S. M.; Troshin, P. A. Energies (Basel, Switz.) 2021, 14, 669. doi:10.3390/en14030669
Return to citation in text: [1] -
Bondy, C. R.; Loeb, S. J. Coord. Chem. Rev. 2003, 240, 77–99. doi:10.1016/s0010-8545(02)00304-1
Return to citation in text: [1] -
Martínez-Crespo, L.; Halgreen, L.; Soares, M.; Marques, I.; Félix, V.; Valkenier, H. Org. Biomol. Chem. 2021, 19, 8324–8337. doi:10.1039/d1ob01279g
Return to citation in text: [1] -
Tomyn, S.; Shylin, S. I.; Bykov, D.; Ksenofontov, V.; Gumienna-Kontecka, E.; Bon, V.; Fritsky, I. O. Nat. Commun. 2017, 8, 14099. doi:10.1038/ncomms14099
Return to citation in text: [1] -
Shylin, S. I.; Pogrebetsky, J. L.; Husak, A. O.; Bykov, D.; Mokhir, A.; Hampel, F.; Shova, S.; Ozarowski, A.; Gumienna-Kontecka, E.; Fritsky, I. O. Chem. Commun. 2021, 57, 11060–11063. doi:10.1039/d1cc04870h
Return to citation in text: [1] -
Semakin, A. N.; Kokuev, A. O.; Nelyubina, Y. V.; Sukhorukov, A. Y.; Zhmurov, P. A.; Ioffe, S. L.; Tartakovsky, V. A. Beilstein J. Org. Chem. 2016, 12, 2471–2477. doi:10.3762/bjoc.12.241
See our previous paper for preliminary result of the cyclization of trishydrazone 3b.
Return to citation in text: [1] [2] [3] -
Semakin, A. N.; Sukhorukov, A. Yu.; Lesiv, A. V.; Khomutova, Y. A.; Ioffe, S. L.; Lyssenko, K. A. Synthesis 2007, 2862–2866. doi:10.1055/s-2007-983847
Return to citation in text: [1] [2] -
Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204
Return to citation in text: [1] -
Churches, Q. I.; Hutton, C. A. Introduction, Interconversion and Removal of Boron Protecting Groups. Boron Reagents in Synthesis; ACS Symposium Series, Vol. 1236; American Chemical Society: Washington, D.C., USA, 2016; pp 357–377. doi:10.1021/bk-2016-1236.ch011
Return to citation in text: [1] -
Golovanov, I. S.; Sukhorukov, A. Y. Top. Curr. Chem. 2021, 379, 8. doi:10.1007/s41061-020-00317-3
Return to citation in text: [1] -
Semakin, A. N.; Golovanov, I. S.; Sukhorukov, A. Y.; Ioffe, S. L.; Tartakovsky, V. A. Russ. Chem. Bull. 2016, 65, 2270–2277. doi:10.1007/s11172-016-1577-7
Return to citation in text: [1] -
Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. J. Appl. Crystallogr. 2021, 54, 1006–1011. doi:10.1107/s1600576721002910
Return to citation in text: [1] -
Kang, Y. K.; Park, H. S. J. Mol. Struct.: THEOCHEM 2004, 676, 171–176. doi:10.1016/j.theochem.2004.01.024
Return to citation in text: [1] -
Cox, C.; Lectka, T. J. Org. Chem. 1998, 63, 2426–2427. doi:10.1021/jo9800863
Return to citation in text: [1] -
Pinalli, R.; Nachtigall, F. F.; Ugozzoli, F.; Dalcanale, E. Angew. Chem., Int. Ed. 1999, 38, 2377–2380. doi:10.1002/(sici)1521-3773(19990816)38:16<2377::aid-anie2377>3.0.co;2-o
Return to citation in text: [1]
| 36. | Semakin, A. N.; Sukhorukov, A. Yu.; Lesiv, A. V.; Khomutova, Y. A.; Ioffe, S. L.; Lyssenko, K. A. Synthesis 2007, 2862–2866. doi:10.1055/s-2007-983847 |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 1. | Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028 |
| 2. | Hu, X.; Meyer, K. J. Organomet. Chem. 2005, 690, 5474–5484. doi:10.1016/j.jorganchem.2005.07.119 |
| 3. | Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308 |
| 8. | Zhang, G.; Mastalerz, M. Chem. Soc. Rev. 2014, 43, 1934–1947. doi:10.1039/c3cs60358j |
| 25. | Golovanov, I. S.; Mazeina, G. S.; Nelyubina, Y. V.; Novikov, R. A.; Mazur, A. S.; Britvin, S. N.; Tartakovsky, V. A.; Ioffe, S. L.; Sukhorukov, A. Y. J. Org. Chem. 2018, 83, 9756–9773. doi:10.1021/acs.joc.8b01296 |
| 42. | Kang, Y. K.; Park, H. S. J. Mol. Struct.: THEOCHEM 2004, 676, 171–176. doi:10.1016/j.theochem.2004.01.024 |
| 43. | Cox, C.; Lectka, T. J. Org. Chem. 1998, 63, 2426–2427. doi:10.1021/jo9800863 |
| 7. | Savyasachi, A. J.; Kotova, O.; Shanmugaraju, S.; Bradberry, S. J.; Ó’Máille, G. M.; Gunnlaugsson, T. Chem 2017, 3, 764–811. doi:10.1016/j.chempr.2017.10.006 |
| 25. | Golovanov, I. S.; Mazeina, G. S.; Nelyubina, Y. V.; Novikov, R. A.; Mazur, A. S.; Britvin, S. N.; Tartakovsky, V. A.; Ioffe, S. L.; Sukhorukov, A. Y. J. Org. Chem. 2018, 83, 9756–9773. doi:10.1021/acs.joc.8b01296 |
| 44. | Pinalli, R.; Nachtigall, F. F.; Ugozzoli, F.; Dalcanale, E. Angew. Chem., Int. Ed. 1999, 38, 2377–2380. doi:10.1002/(sici)1521-3773(19990816)38:16<2377::aid-anie2377>3.0.co;2-o |
| 6. | Valášek, M.; Lindner, M.; Mayor, M. Beilstein J. Nanotechnol. 2016, 7, 374–405. doi:10.3762/bjnano.7.34 |
| 24. | Golovanov, I. S.; Sukhorukov, A. Y.; Nelyubina, Y. V.; Khomutova, Y. A.; Ioffe, S. L.; Tartakovsky, V. A. J. Org. Chem. 2015, 80, 6728–6736. doi:10.1021/acs.joc.5b00892 |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 4. | Kuswandi, B.; Nuriman; Verboom, W.; Reinhoudt, D. N. Sensors 2006, 6, 978–1017. doi:10.3390/s6080978 |
| 5. | Wadas, T. J.; Wong, E. H.; Weisman, G. R.; Anderson, C. J. Chem. Rev. 2010, 110, 2858–2902. doi:10.1021/cr900325h |
| 25. | Golovanov, I. S.; Mazeina, G. S.; Nelyubina, Y. V.; Novikov, R. A.; Mazur, A. S.; Britvin, S. N.; Tartakovsky, V. A.; Ioffe, S. L.; Sukhorukov, A. Y. J. Org. Chem. 2018, 83, 9756–9773. doi:10.1021/acs.joc.8b01296 |
| 26. | de Vries, R. H.; Viel, J. H.; Kuipers, O. P.; Roelfes, G. Angew. Chem., Int. Ed. 2021, 60, 3946–3950. doi:10.1002/anie.202011460 |
| 41. | Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. J. Appl. Crystallogr. 2021, 54, 1006–1011. doi:10.1107/s1600576721002910 |
| 10. | Fleck, C.; Franzmann, E.; Claes, D.; Rickert, A.; Maison, W. Synthesis 2013, 45, 1452–1461. doi:10.1055/s-0033-1338470 |
| 11. | Grillaud, M.; Bianco, A. J. Pept. Sci. 2015, 21, 330–345. doi:10.1002/psc.2719 |
| 12. | Lamanna, G.; Grillaud, M.; Macri, C.; Chaloin, O.; Muller, S.; Bianco, A. Biomaterials 2014, 35, 7553–7561. doi:10.1016/j.biomaterials.2014.05.017 |
| 13. | Fleck, C.; Memmel, E.; Fölsing, M.; Poll, B.; Hackl, T.; Seibel, J.; Maison, W. Eur. J. Org. Chem. 2015, 1696–1710. doi:10.1002/ejoc.201403517 |
| 15. | Balija, A. M.; Kohman, R. E.; Zimmerman, S. C. Angew. Chem., Int. Ed. 2008, 47, 8072–8074. doi:10.1002/anie.200802222 |
| 16. | Kohman, R. E.; Zimmerman, S. C. Chem. Commun. 2009, 794–796. doi:10.1039/b818183g |
| 17. | Hou, T.; Zhang, J.; Wang, C.; Luo, J. Org. Chem. Front. 2017, 4, 1819–1823. doi:10.1039/c7qo00357a |
| 18. | Semakin, A. N.; Nelyubina, Y. V.; Ioffe, S. L.; Sukhorukov, A. Y. Eur. J. Org. Chem. 2020, 6723–6735. doi:10.1002/ejoc.202000832 |
| 19. | Suslov, E. V.; Ponomarev, K. Y.; Volcho, K. P.; Salakhutdinov, N. F. Russ. J. Bioorg. Chem. 2021, 47, 1133–1154. doi:10.1134/s1068162021060236 |
| 20. | Dalinger, A. I.; Medved’ko, A. V.; Kalinin, M. A.; Sereda, V. A.; Churakov, A. V.; Vatsadze, S. Z. Russ. Chem. Bull. 2021, 70, 1002–1005. doi:10.1007/s11172-021-3180-9 |
| 35. |
Semakin, A. N.; Kokuev, A. O.; Nelyubina, Y. V.; Sukhorukov, A. Y.; Zhmurov, P. A.; Ioffe, S. L.; Tartakovsky, V. A. Beilstein J. Org. Chem. 2016, 12, 2471–2477. doi:10.3762/bjoc.12.241
See our previous paper for preliminary result of the cyclization of trishydrazone 3b. |
| 4. | Kuswandi, B.; Nuriman; Verboom, W.; Reinhoudt, D. N. Sensors 2006, 6, 978–1017. doi:10.3390/s6080978 |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 22. | Semakin, A. N.; Sukhorukov, A. Y.; Nelyubina, Y. V.; Khomutova, Y. A.; Ioffe, S. L.; Tartakovsky, V. A. J. Org. Chem. 2014, 79, 6079–6086. doi:10.1021/jo5007703 |
| 23. | Semakin, A. N.; Sukhorukov, A. Y. Targets Heterocycl. Syst. 2017, 21, 82–97. |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 2. | Hu, X.; Meyer, K. J. Organomet. Chem. 2005, 690, 5474–5484. doi:10.1016/j.jorganchem.2005.07.119 |
| 4. | Kuswandi, B.; Nuriman; Verboom, W.; Reinhoudt, D. N. Sensors 2006, 6, 978–1017. doi:10.3390/s6080978 |
| 37. | Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204 |
| 38. | Churches, Q. I.; Hutton, C. A. Introduction, Interconversion and Removal of Boron Protecting Groups. Boron Reagents in Synthesis; ACS Symposium Series, Vol. 1236; American Chemical Society: Washington, D.C., USA, 2016; pp 357–377. doi:10.1021/bk-2016-1236.ch011 |
| 39. | Golovanov, I. S.; Sukhorukov, A. Y. Top. Curr. Chem. 2021, 379, 8. doi:10.1007/s41061-020-00317-3 |
| 9. | Beuerle, F.; Gole, B. Angew. Chem., Int. Ed. 2018, 57, 4850–4878. doi:10.1002/anie.201710190 |
| 10. | Fleck, C.; Franzmann, E.; Claes, D.; Rickert, A.; Maison, W. Synthesis 2013, 45, 1452–1461. doi:10.1055/s-0033-1338470 |
| 14. | Ivleva, E. A.; Pogulyaiko, A. V.; Klimochkin, Y. N. Russ. J. Org. Chem. 2018, 54, 1294–1300. doi:10.1134/s107042801809004x |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 40. | Semakin, A. N.; Golovanov, I. S.; Sukhorukov, A. Y.; Ioffe, S. L.; Tartakovsky, V. A. Russ. Chem. Bull. 2016, 65, 2270–2277. doi:10.1007/s11172-016-1577-7 |
| 30. | Ozerova, V. V.; Zhidkov, I. S.; Boldyreva, A.; Dremova, N. N.; Emelianov, N. A.; Shilov, G. V.; Frolova, L. A.; Kurmaev, E. Z.; Sukhorukov, A. Y.; Aldoshin, S. M.; Troshin, P. A. Energies (Basel, Switz.) 2021, 14, 669. doi:10.3390/en14030669 |
| 25. | Golovanov, I. S.; Mazeina, G. S.; Nelyubina, Y. V.; Novikov, R. A.; Mazur, A. S.; Britvin, S. N.; Tartakovsky, V. A.; Ioffe, S. L.; Sukhorukov, A. Y. J. Org. Chem. 2018, 83, 9756–9773. doi:10.1021/acs.joc.8b01296 |
| 27. | Samoľová, E.; Premužić, D.; Plociennik, S.; Hołyńska, M. J. Mol. Struct. 2019, 1176, 366–375. doi:10.1016/j.molstruc.2018.08.100 |
| 28. | Premužić, D.; Hołyńska, M.; Ozarowski, A.; Pietzonka, C.; Roseborough, A.; Stoian, S. A. Inorg. Chem. 2020, 59, 10768–10784. doi:10.1021/acs.inorgchem.0c01242 |
| 29. | Golovanov, I. S.; Leonov, A. V.; Lesnikov, V. K.; Pospelov, E. V.; Frolov, K. V.; Korlyukov, A. A.; Nelyubina, Y. V.; Novikov, V. V.; Sukhorukov, A. Y. Dalton Trans. 2022, 51, 4284–4296. doi:10.1039/d1dt04104e |
| 35. |
Semakin, A. N.; Kokuev, A. O.; Nelyubina, Y. V.; Sukhorukov, A. Y.; Zhmurov, P. A.; Ioffe, S. L.; Tartakovsky, V. A. Beilstein J. Org. Chem. 2016, 12, 2471–2477. doi:10.3762/bjoc.12.241
See our previous paper for preliminary result of the cyclization of trishydrazone 3b. |
| 36. | Semakin, A. N.; Sukhorukov, A. Yu.; Lesiv, A. V.; Khomutova, Y. A.; Ioffe, S. L.; Lyssenko, K. A. Synthesis 2007, 2862–2866. doi:10.1055/s-2007-983847 |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 18. | Semakin, A. N.; Nelyubina, Y. V.; Ioffe, S. L.; Sukhorukov, A. Y. Eur. J. Org. Chem. 2020, 6723–6735. doi:10.1002/ejoc.202000832 |
| 33. | Tomyn, S.; Shylin, S. I.; Bykov, D.; Ksenofontov, V.; Gumienna-Kontecka, E.; Bon, V.; Fritsky, I. O. Nat. Commun. 2017, 8, 14099. doi:10.1038/ncomms14099 |
| 34. | Shylin, S. I.; Pogrebetsky, J. L.; Husak, A. O.; Bykov, D.; Mokhir, A.; Hampel, F.; Shova, S.; Ozarowski, A.; Gumienna-Kontecka, E.; Fritsky, I. O. Chem. Commun. 2021, 57, 11060–11063. doi:10.1039/d1cc04870h |
| 35. |
Semakin, A. N.; Kokuev, A. O.; Nelyubina, Y. V.; Sukhorukov, A. Y.; Zhmurov, P. A.; Ioffe, S. L.; Tartakovsky, V. A. Beilstein J. Org. Chem. 2016, 12, 2471–2477. doi:10.3762/bjoc.12.241
See our previous paper for preliminary result of the cyclization of trishydrazone 3b. |
| 21. | Semakin, A. N.; Sukhorukov, A. Y.; Lesiv, A. V.; Ioffe, S. L.; Lyssenko, K. A.; Nelyubina, Y. V.; Tartakovsky, V. A. Org. Lett. 2009, 11, 4072–4075. doi:10.1021/ol9015157 |
| 31. | Bondy, C. R.; Loeb, S. J. Coord. Chem. Rev. 2003, 240, 77–99. doi:10.1016/s0010-8545(02)00304-1 |
| 32. | Martínez-Crespo, L.; Halgreen, L.; Soares, M.; Marques, I.; Félix, V.; Valkenier, H. Org. Biomol. Chem. 2021, 19, 8324–8337. doi:10.1039/d1ob01279g |
© 2022 Semakin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.