Abstract
The total synthesis of (3R,5R)-harzialactone A (1) and its (3R,5S)-isomer (2) is described. Epoxide opening with thioacetal and diastereoselective reductions are used as key reactions.

Graphical Abstract
Introduction
Marine microorganisms such as bacteria, fungi, and microalgae have proved to be a rich source of structurally novel and biologically active secondary metabolites [1]. (+)-Harzialactone A (1), a marine metabolite isolated from the culture broth of a strain of Trichoderma harzianum OUPS-N115 by Numata and co-workers, exhibited antitumor and cytotoxic activities against cultured P388 cells [2]. The absolute configuration of (+)-1 was established based on 1H NMR studies and by its synthesis [3,4]. Harzialactone A (1) (Figure 1) is a synthetic target of considerable interest due to its potent biological activity and unique structure. A few methods for its synthesis have been documented in the literature [3-10] as well as a synthesis of nonnatural (−)-harzialactone A [11]. However, the anti-tumor activity of Harzialactone A coupled with its unique structural architecture prompted us to attempt its synthesis.
![[1860-5397-6-8-1]](/bjoc/content/figures/1860-5397-6-8-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
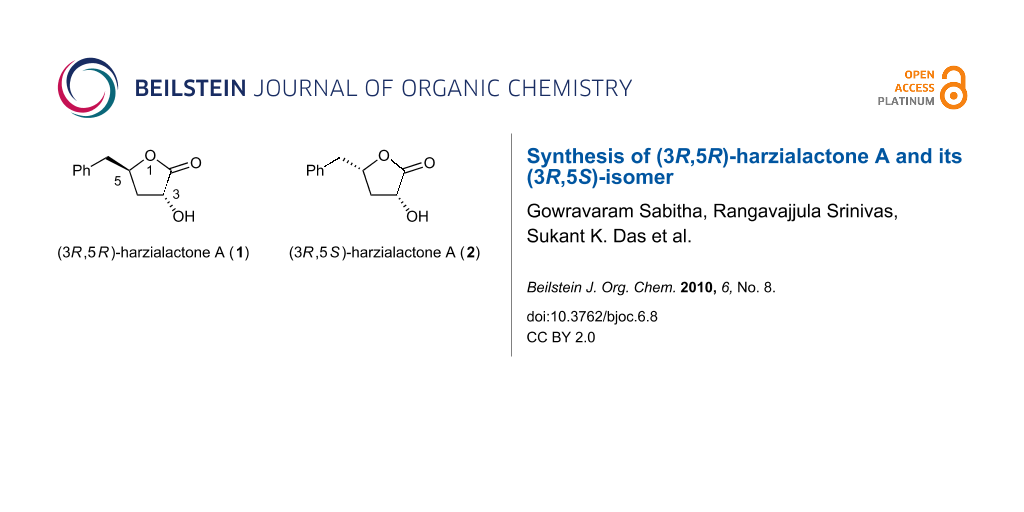
Figure 1: Natural harzialactone A (1), and its (3R,5S)-isomer (2).
Figure 1: Natural harzialactone A (1), and its (3R,5S)-isomer (2).
The retrosynthesis is depicted in Scheme 1. Harzialactone 1 could be made from 3 by successive protecting group transformations. 3 can be made by hydroxyl directed reduction of 4 which in turn could be prepared by epoxide 6 opening with dithiane 5.
![[1860-5397-6-8-i1]](/bjoc/content/inline/1860-5397-6-8-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of harzialactone A (1).
Scheme 1: Retrosynthesis of harzialactone A (1).
Results and Discussion
The synthesis of natural (3R,5R)-1 was initiated from the known epoxide 6, which is commercially available. Treatment of 2-phenylacetaldehyde 7 with 1,3-propanedithiol in the presence of BF3·Et2O in CH2Cl2 afforded thioacetal 5 in 90% yield (Scheme 2). The epoxide 6 was coupled with the acyl anion equivalent 5 (1.0 equiv), prepared by metallation at –78 °C with 1.0 equiv of n-butyllithium in the presence of BF3·Et2O to obtain 8 in 64% yield. Removal of the dithioketal using HgCl2/CaCO3 in CH3CN/H2O (4:1)[12] provided the corresponding hydroxyketone 4 in 82% yield. Treatment of 5 with NaBH4 and MeOBEt2 [13,14] stereoselectively formed the syn diol 9 in good yield (80%). The diol 9 was subsequently transformed into the isopropylidene derivative 3 by treatment with 2,2-dimethoxypropane and a catalytic amount of PPTS in CH2Cl2.
![[1860-5397-6-8-i2]](/bjoc/content/inline/1860-5397-6-8-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of natural harzialactone A (1).
Scheme 2: Synthesis of natural harzialactone A (1).
In the 13C NMR spectrum of 3, the acetonide methyl groups resonated at 19.6 and 29.9 ppm indicating a 1,3-syn-relationship that was further substantiated by the appearance of the quaternary carbon in the downfield region (98.7 ppm). Deprotection of the benzyl group using Li/liq. NH3 gave alcohol 10. Oxidation of alcohol 10 under Swern conditions and further oxidation of the resulting aldehyde using NaH2PO4, NaClO2 in DMSO/H2O furnished the target hydroxylactone (3R,5R)-1 as reported earlier. The IR absorption at 1774 cm−1 indicates the presence of δ-lactone system.
The synthesis of (3R,5S)-2 was also accomplished in an identical manner from 4 (Scheme 3). The substrate hydroxyl directed asymmetric reduction with Me4NBH(OAc)3 [15,16] was performed at 0 °C to afford the anti diol 11 as the major product, which was converted into stereoisomer (3R,5S)-2 via acetonide 12, deprotection of benzyl group to give 13, and further functional group transformations by use of the same reagents and conditions as those described for the conversion of 10 into 1. The IR absorption at 1775 cm−1 confirms the presence of δ-lactone in (3R,5S)-2.
![[1860-5397-6-8-i3]](/bjoc/content/inline/1860-5397-6-8-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of (3R,5S)-harzialactone A (2).
Scheme 3: Synthesis of (3R,5S)-harzialactone A (2).
The anti relationship of two hydroxyl groups was studied in compound 12. In the 13C NMR of 12, the acetonide methyl groups resonated at 24.9 and 34.2 ppm indicating a 1,3-anti-relationship that was further substantiated by the appearance of the quaternary carbon in the downfield region (100.5 ppm) [7].
In conclusion, a stereoselective synthesis of natural (+)-(3R,5R)-harzialactone A and its nonnatural stereoisomer (3R,5S) has been accomplished.
Supporting Information
| Supporting Information File 1: Experimental section and analytical data. | ||
| Format: DOC | Size: 4.4 MB | Download |
References
-
Blunt, J. W.; Copp, B. R.; Munro, M. H. G.; Northcote, P. T.; Prinsep, P. R. Nat. Prod. Rep. 2004, 21, 1–49. doi:10.1039/b305250h
(and references cited therein).
Return to citation in text: [1] -
Amagata, T.; Usami, Y.; Minoura, K.; Ito, T.; Numata, A. J. Antibiot. 1998, 51, 33–40.
Return to citation in text: [1] -
Mereyala, H. B.; Gadikota, R. R. Tetrahedron: Asymmetry 1999, 10, 2305–2306. doi:10.1016/S0957-4166(99)00245-1
Return to citation in text: [1] [2] -
Mereyala, H. B.; Joe, M.; Gadikota, R. R. Tetrahedron: Asymmetry 2000, 11, 4071–4081. doi:10.1016/S0957-4166(00)00389-X
Return to citation in text: [1] [2] -
Ikota, N. Heterocycles 1991, 32, 521–528. doi:10.3987/COM-91-5669
Return to citation in text: [1] -
Kumar, J. S. R.; Datta, A. Tetrahedron Lett. 1999, 40, 1381–1383. doi:10.1016/S0040-4039(98)02614-8
Return to citation in text: [1] -
Kiyooka, S.; Goh, K.; Nakamura, Y.; Takesue, H.; Hena, M. A. Tetrahedron Lett. 2000, 41, 6599–6603. doi:10.1016/S0040-4039(00)01124-2
Return to citation in text: [1] [2] -
Moreau, X.; Campagne, J. Tetrahedron Lett. 2001, 42, 4467–4469. doi:10.1016/S0040-4039(01)00753-5
Return to citation in text: [1] -
Kotkar, S. P.; Suryavanshi, G. S.; Sudalai, A. Tetrahedron: Asymmetry 2007, 18, 1795–1798. doi:10.1016/j.tetasy.2007.07.031
Return to citation in text: [1] -
Kumar, A. N.; Bhatt, S.; Chattopadhyay, S. Tetrahedron: Asymmetry 2009, 20, 205–209. doi:10.1016/j.tetasy.2009.01.009
Return to citation in text: [1] -
Jian, Y.-J.; Wu, Y.; Li, L.; Lu, J. Tetrahedron: Asymmetry 2005, 16, 2649–2651. doi:10.1016/j.tetasy.2005.07.003
Return to citation in text: [1] -
Corey, E. J.; Bock, M. G. Tetrahedron Lett. 1975, 16, 2643–2646. doi:10.1016/S0040-4039(00)75203-8
Return to citation in text: [1] -
Hanamoto, T.; Hiyama, T. Tetrahedron Lett. 1988, 29, 6467–6470. doi:10.1016/S0040-4039(00)82375-8
Return to citation in text: [1] -
Chen, K. M.; Hardtmanna, G. E.; Prasad, K.; Pepc, O.; Shapinro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/S0040-4039(00)95673-9
Return to citation in text: [1] -
Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035
Return to citation in text: [1] -
Evans, D. A.; Chapman, K. T. Tetrahedron Lett. 1986, 27, 5939–5942. doi:10.1016/S0040-4039(00)85367-8
Return to citation in text: [1]
| 1. |
Blunt, J. W.; Copp, B. R.; Munro, M. H. G.; Northcote, P. T.; Prinsep, P. R. Nat. Prod. Rep. 2004, 21, 1–49. doi:10.1039/b305250h
(and references cited therein). |
| 11. | Jian, Y.-J.; Wu, Y.; Li, L.; Lu, J. Tetrahedron: Asymmetry 2005, 16, 2649–2651. doi:10.1016/j.tetasy.2005.07.003 |
| 3. | Mereyala, H. B.; Gadikota, R. R. Tetrahedron: Asymmetry 1999, 10, 2305–2306. doi:10.1016/S0957-4166(99)00245-1 |
| 4. | Mereyala, H. B.; Joe, M.; Gadikota, R. R. Tetrahedron: Asymmetry 2000, 11, 4071–4081. doi:10.1016/S0957-4166(00)00389-X |
| 5. | Ikota, N. Heterocycles 1991, 32, 521–528. doi:10.3987/COM-91-5669 |
| 6. | Kumar, J. S. R.; Datta, A. Tetrahedron Lett. 1999, 40, 1381–1383. doi:10.1016/S0040-4039(98)02614-8 |
| 7. | Kiyooka, S.; Goh, K.; Nakamura, Y.; Takesue, H.; Hena, M. A. Tetrahedron Lett. 2000, 41, 6599–6603. doi:10.1016/S0040-4039(00)01124-2 |
| 8. | Moreau, X.; Campagne, J. Tetrahedron Lett. 2001, 42, 4467–4469. doi:10.1016/S0040-4039(01)00753-5 |
| 9. | Kotkar, S. P.; Suryavanshi, G. S.; Sudalai, A. Tetrahedron: Asymmetry 2007, 18, 1795–1798. doi:10.1016/j.tetasy.2007.07.031 |
| 10. | Kumar, A. N.; Bhatt, S.; Chattopadhyay, S. Tetrahedron: Asymmetry 2009, 20, 205–209. doi:10.1016/j.tetasy.2009.01.009 |
| 3. | Mereyala, H. B.; Gadikota, R. R. Tetrahedron: Asymmetry 1999, 10, 2305–2306. doi:10.1016/S0957-4166(99)00245-1 |
| 4. | Mereyala, H. B.; Joe, M.; Gadikota, R. R. Tetrahedron: Asymmetry 2000, 11, 4071–4081. doi:10.1016/S0957-4166(00)00389-X |
| 2. | Amagata, T.; Usami, Y.; Minoura, K.; Ito, T.; Numata, A. J. Antibiot. 1998, 51, 33–40. |
| 7. | Kiyooka, S.; Goh, K.; Nakamura, Y.; Takesue, H.; Hena, M. A. Tetrahedron Lett. 2000, 41, 6599–6603. doi:10.1016/S0040-4039(00)01124-2 |
| 15. | Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035 |
| 16. | Evans, D. A.; Chapman, K. T. Tetrahedron Lett. 1986, 27, 5939–5942. doi:10.1016/S0040-4039(00)85367-8 |
| 13. | Hanamoto, T.; Hiyama, T. Tetrahedron Lett. 1988, 29, 6467–6470. doi:10.1016/S0040-4039(00)82375-8 |
| 14. | Chen, K. M.; Hardtmanna, G. E.; Prasad, K.; Pepc, O.; Shapinro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/S0040-4039(00)95673-9 |
| 12. | Corey, E. J.; Bock, M. G. Tetrahedron Lett. 1975, 16, 2643–2646. doi:10.1016/S0040-4039(00)75203-8 |
© 2010 Sabitha et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)