Abstract
The synthesis of a number of aminoethyl glycosides of cell-surface carbohydrates, which are important intermediates for glycoarray synthesis, is described. A set of protocols was developed which provide these intermediates, in a short number of steps, from commercially available starting materials.

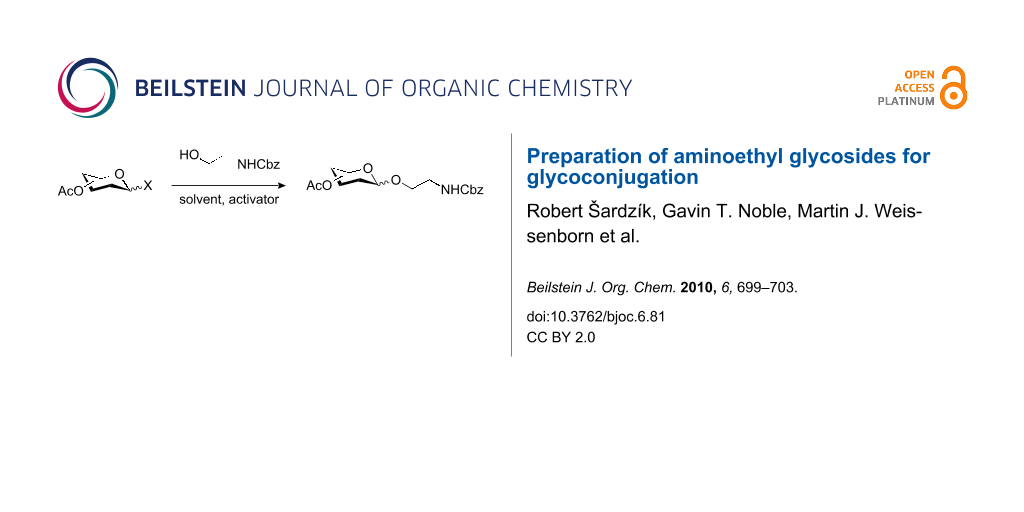
Graphical Abstract
Introduction
The chemical conjugation of carbohydrates through the anomeric centre to biomolecules such as peptides, proteins, lipids, metabolites and to array surfaces is an important synthetic challenge [1-5]. A diverse range of linkers and spacers has been described in the literature [2-12], among which aminoalkyl glycosides have become the most popular, in particular the aminoethyl linker. This linker has been tested in a large number of arrays and appears to be biocompatible in array screening [2,3,7]. Given that aminoethyl glycosides are conveniently conjugated to surfaces containing activated carboxylates, they have become a useful generic anomeric functional group for glycoconjugation. The importance of this linker merits efforts into finding a robust synthetic method than can be used by scientists who are not experienced in carbohydrate synthesis. Here we describe a systematic study with the aim of finding such robust and efficient methods for a number of commonly used mono- and disaccharides starting from commercially available reagents and with a minimal number of steps. In our studies no such general aminoethylation method that was applicable to all targets was found, which rather suggests that protocols need to be tailored for each sugar.
Results and Discussion
Coupling reactions
Aminoethyl glycosides have previously been generated in a number of ways. Free sugars have been glycosylated with 2-chloroethanol under acid catalysis, followed by peracetylation, nucleophilic substitution with azide and finally, reduction of the azido group [13,14]. Alternatively, the carbohydrate was first activated as the trichloroacetimidate or bromide followed by glycosylation with N-Cbz-aminoethanol [15], bromoethanol [16] or azidoethanol [17] and subsequently transformed into the amine.
In the interest of finding fast reliable methods, we have investigated two general aminoethylation protocols: First, the direct glycosylation of peracetylated sugars, which can be either purchased or easily prepared from free sugars and can be used without purification. Where these proved to be unreactive, the anomeric acetates were converted to glycosyl bromides, usually in quantitative yields, and products were used without further purification. Where possible, N-Cbz-protected aminoethanol was used as the glycosyl acceptor because it is commercially available, crystalline and can be easily deprotected in one step avoiding use of azides. Figure 1 lists the target aminoethyl glycosides (1–9) generated in this study (q.v. Scheme 1 and Table 1).
![[1860-5397-6-81-1]](/bjoc/content/figures/1860-5397-6-81-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Aminoethyl glycosides (1–9) which were synthesised in this study.
Figure 1: Aminoethyl glycosides (1–9) which were synthesised in this study.
The key glycosylation step is shown in Scheme 1 and the results of the different glycosylation reactions are summarised in Table 1.
![[1860-5397-6-81-i1]](/bjoc/content/inline/1860-5397-6-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General reaction scheme for generation of aminoethyl glycosides. X = OAc, Br or Cl.
Scheme 1: General reaction scheme for generation of aminoethyl glycosides. X = OAc, Br or Cl.
Table 1: Results of glycosylation reactions as shown in Scheme 1.
| Entry | Product | X | Method | α/βc | Yieldd |
|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-81-i3.svg?max-width=637&scale=1.0)
10 |
Br | A | 17:83 | 67 + 23e |
| 2 | 10 | Br | B | 23:77 | 58 + 19e |
| 3 | 10 | OAc | C | 22:78 | 52 + 17e |
| 4 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-81-i4.svg?max-width=637&scale=1.0)
11 |
OAc | C | 15:85 | 36 |
| 5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-81-i5.svg?max-width=637&scale=1.0)
12 |
OAc | C | 8:92 | 62 |
| 6 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-81-i6.svg?max-width=637&scale=1.0)
13 |
OAc | C | >95:5 | 57 |
| 7 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-81-i7.svg?max-width=637&scale=1.0)
14 |
Br | A | 14:86 | 75 + 19e |
| 8 | 14 | Br | B | 15:85 | 75 + 20e |
| 9 | 14 | OAc | C | 35:65 | n.d. |
| 10 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-81-i8.svg?max-width=637&scale=1.0)
15 |
OAc | D | >5:95 | 61 |
| 11 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-81-i9.svg?max-width=637&scale=1.0)
16 |
Br | A | 10:90 | 56 |
| 12 | 16 | Br | B | 10:90 | 86 |
| 13 | 16 | Br | Aa | 31:68 | 30 |
| 14 | 16 | Br | Bb | 30:70 | 73e |
| 15 | 16 | Br | E | 37:63 | 59 |
| 16 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-6-81-i10.svg?max-width=637&scale=1.0)
17 |
Br | E | 10:90 | 88 |
| 17 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-6-81-i11.svg?max-width=637&scale=1.0)
18 |
Br | B | 14:86 | 47 |
| 18 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-6-81-i12.svg?max-width=637&scale=1.0)
19 |
Cl | F | 10:90 | 70 |
aThe reaction was performed overnight at r.t. bThe reaction was performed in CH2Cl2. cdetermined by 13C NMR from crude reaction mixture. dYield of pure major anomer (>95:5) after column chromatography. eMixture of both anomers. Method A: Hg(CN)2, CH3CN, 60 °C, 2–4 h. Method B: Hg(CN)2, CH3CN, 90 °C, microwave, 200 W, 15 min. Method C: BF3·Et2O, CH3CN, 0 °C, 1 h, r.t., overnight. Method D: SnCl4, CH3CN, 60 °C, 16 h. Method E: Hg(CN)2, HgBr2, CH3CN, r.t., overnight. Method F: Ag2CO3, CH2Cl2, r.t., overnight.
Fully protected xylopyranoside 10 could be prepared both from the corresponding bromide as well as the acetate (the β-anomer was prepared from xylose with sodium acetate in acetic anhydride) in similar yields. In each case both anomers were formed with moderate selectivity and from the reaction mixture the pure β-anomer (>95:5) was isolated by column chromatography.
The glucoside 11, galactoside 12 and mannoside 13 were prepared in moderate to good yield directly from the acetate (X = OAc in Scheme 1) giving rapid access to these monosaccharide derivatives.
The fucoside 14 was generated both from the acetate and bromide. Higher alpha selectivity was observed with the bromide (α/β ratio of crude product 82:18).
N-Acetylglucosamine 15 was successfully prepared from the β-acetate using SnCl4 (method D). When starting from the α-acetate, no reaction was observed and only starting material could be recovered. Under microwave conditions (method B or using other Lewis acids as Yb(OTf)3 in DCM, 90 °C, 30 min, 200 W) the reaction was not reproducible, giving low yields and leading to decomposition products.
Lactose is both cheap and readily available. It is an important component of glycoprotein glycans and also a substrate for sialyltransferases to generate biologically important sialyllactosides. The aminoethyl lactoside 16 was prepared in greatest yield from the bromide and attempts to prepare 16 directly from the acetate using BF3·Et2O as the activator only resulted in decomposition.
A number of reaction conditions for generating 16 from the bromide with N-Cbz-aminoethanol were investigated. With Ag2CO3 in dichloromethane at room temperature low yields of product 16 along with a number of side-products (orthoester, elimination or hydrolysis) were observed and the product was difficult to separate from starting materials, in particular the N-Cbz-aminoethanol. With Hg(CN)2, or the more reactive Hg(CN)2/HgBr2-mixture, in dichloromethane or acetonitrile, glycosylation was more successful, but both anomers were generated. Given the problems previously encountered with purification, the glycosylation with Hg(CN)2 was further optimised by increasing both the temperature and the amount of acceptor. The problem of separation of the alcohol from the product was solved by acetylation of the crude reaction mixture to lower the polarity of the free alcohol. Attempts to speed up the reaction by heating led to the observation that in acetonitrile predominantly one (β) anomer is formed, but anomerisation occurs with longer reaction times. In dichloromethane both anomers were formed. The best reaction conditions were combined to give Method A. The success of Method A led to attempts to improve the method further and to use microwave irradiation as in method B. Method B also works well for monosaccharides and maltoside.
Given the problems with purification, the use of azidoethanol as a glycosyl acceptor was also investigated. This reaction (Table 1) was much more successful and produced mainly the beta anomer 17.
Maltoside 18 was generated by the same microwave-mediated glycosylation as developed for lactoside 16 (Method B) and in reasonable yield.
N-Acetyl neuraminic acid (sialic acid) is an important component of cell surfaces and chemical glycosylation procedures involving sialic acid are generally challenging. In our hands activation as the chloride (prepared from Neu5Ac in 3 steps) using silver carbonate (Method F) gave reasonable yields of 19.
Deprotection reactions
The general deprotection for compounds 10–18 is shown in Scheme 2. Acetates were cleaved using NaOMe followed by hydrogenation to generate 1–8 in good yields.
![[1860-5397-6-81-i2]](/bjoc/content/inline/1860-5397-6-81-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Deprotection was also successful when the hydrogenation was performed first, but in some cases migration of acetate to the aminoethyl linker was observed. However, this can be avoided by using palladium hydroxide on charcoal as the hydrogenation catalyst, with short reaction times, followed by the immediate use of the resulting amine in further coupling [18].
Sialoside 19 was deprotected by treatment with NaOMe, followed by LiOH and subsequent hydrogenation to give 9.
Conclusion
We have described rapid and convenient methods for the synthesis of a range of aminoethyl glycosides (1–9) of common mono- and disaccharides. Although some of the glycosylation reactions could be improved by using alternative glycosylation methods (such as trichloroacetimidates, thiols), these would require more steps with chromatographic purifications and less overall yields. These aminoethyl glycosides are now readily accessible for incorporation into glycan arrays.
Supporting Information
A Supporting Information containing all experimental details and analytical data of all compounds described in the article as well as their precursors is available.
| Supporting Information File 1: Experimental procedures and analytical data | ||
| Format: PDF | Size: 661.2 KB | Download |
References
-
Lepenies, B.; Seeberger, P. H. Immunopharmacol. Immunotoxicol. 2010, 32, 196–207. doi:10.3109/08923970903292663
Return to citation in text: [1] -
Blixt, O.; Han, S.; Liao, L.; Zeng, Y.; Hoffmann, J.; Futakawa, S.; Paulson, J. C. J. Am. Chem. Soc. 2008, 130, 6680–6681. doi:10.1021/ja801052g
Return to citation in text: [1] [2] [3] -
Huflejt, M. E.; Vuskovic, M.; Vasiliu, D.; Xu, H.; Obukhova, P.; Shilova, N.; Tuzikov, A.; Galanina, O.; Arun, B.; Lu, K.; Bovin, N. Mol. Immunol. 2009, 46, 3037–3049. doi:10.1016/j.molimm.2009.06.010
Return to citation in text: [1] [2] [3] -
Bejugam, M.; Flitsch, S. L. Org. Lett. 2004, 6, 4001–4004. doi:10.1021/ol048342n
Return to citation in text: [1] [2] -
Macmillan, D.; Daines, A. M.; Bayrhuber, M.; Flitsch, S. L. Org. Lett. 2002, 4, 1467–1470. doi:10.1021/ol025627w
Return to citation in text: [1] [2] -
Hartmann, M.; Horst, A. K.; Klemm, P.; Lindhorst, T. K. Chem. Commun. 2010, 46, 330–332. doi:10.1039/b922525k
Return to citation in text: [1] -
Zhi, Z.; Laurent, N.; Powell, A. K.; Karamanska, R.; Fais, M.; Voglmeir, J.; Wright, A.; Blackburn, J. M.; Crocker, P. R.; Russell, D. A.; Flitsch, S. L.; Field, R. A.; Turnbull, J. E. ChemBioChem 2008, 9, 1568–1575. doi:10.1002/cbic.200700788
Return to citation in text: [1] [2] -
Karamanska, R.; Clarke, J.; Blixt, O.; MacRae, J. I.; Zhang, J. Q.; Crocker, P. R.; Laurent, N.; Wright, A.; Flitsch, S. L.; Russell, D. A.; Field, R. A. Glycoconjugate J. 2008, 25, 69–74. doi:10.1007/s10719-007-9047-y
Return to citation in text: [1] -
Laurent, N.; Voglmeir, J.; Wright, A.; Blackburn, J.; Pham, N. T.; Wong, S. C. C.; Gaskell, S. J.; Flitsch, S. L. ChemBioChem 2008, 9, 883–887. doi:10.1002/cbic.200700692
Return to citation in text: [1] -
Laurent, N.; Haddoub, R.; Flitsch, S. L. Trends Biotechnol. 2008, 26, 328–337. doi:10.1016/j.tibtech.2008.03.003
Return to citation in text: [1] -
Laurent, N.; Voglmeir, J.; Flitsch, S. L. Chem. Commun. 2008, 4400–4412. doi:10.1039/b806983m
Return to citation in text: [1] -
Laurent, N.; Haddoub, R.; Voglmeir, J.; Wong, S. C. C.; Gaskell, S. J.; Flitsch, S. L. ChemBioChem 2008, 9, 2592–2596. doi:10.1002/cbic.200800481
Return to citation in text: [1] -
Ni, J.; Singh, S.; Wang, L.-X. Bioconjugate Chem. 2003, 14, 232–238. doi:10.1021/bc025617f
Return to citation in text: [1] -
Sanki, A. K.; Mahal, L. K. Synlett 2006, 455–459. doi:10.1055/s-2006-926264
Return to citation in text: [1] -
Orlandi, S.; Annuziata, R.; Benaglia, M.; Cozzi, F.; Manzoni, L. Tetrahedron 2005, 61, 10048–10060. doi:10.1016/j.tet.2005.08.018
Return to citation in text: [1] -
Park, S.; Shin, I. Org. Lett. 2007, 9, 1675–1678. doi:10.1021/ol070250l
Return to citation in text: [1] -
Chernyak, A. Y.; Sharma, G. V. M.; Kononov, L. O.; Krishna, P. R.; Levinsky, A. B.; Kochetkov, N. K.; Rama Rao, A. V. Carbohydr. Res. 1992, 223, 303–309. doi:10.1016/0008-6215(92)80029-Z
Return to citation in text: [1] -
Noble, G. T.; Flitsch, S. L.; Liem, K. P.; Webb, S. J. Org. Biomol. Chem. 2009, 7, 5245–5254. doi:10.1039/b910976e
Return to citation in text: [1]
| 1. | Lepenies, B.; Seeberger, P. H. Immunopharmacol. Immunotoxicol. 2010, 32, 196–207. doi:10.3109/08923970903292663 |
| 2. | Blixt, O.; Han, S.; Liao, L.; Zeng, Y.; Hoffmann, J.; Futakawa, S.; Paulson, J. C. J. Am. Chem. Soc. 2008, 130, 6680–6681. doi:10.1021/ja801052g |
| 3. | Huflejt, M. E.; Vuskovic, M.; Vasiliu, D.; Xu, H.; Obukhova, P.; Shilova, N.; Tuzikov, A.; Galanina, O.; Arun, B.; Lu, K.; Bovin, N. Mol. Immunol. 2009, 46, 3037–3049. doi:10.1016/j.molimm.2009.06.010 |
| 4. | Bejugam, M.; Flitsch, S. L. Org. Lett. 2004, 6, 4001–4004. doi:10.1021/ol048342n |
| 5. | Macmillan, D.; Daines, A. M.; Bayrhuber, M.; Flitsch, S. L. Org. Lett. 2002, 4, 1467–1470. doi:10.1021/ol025627w |
| 15. | Orlandi, S.; Annuziata, R.; Benaglia, M.; Cozzi, F.; Manzoni, L. Tetrahedron 2005, 61, 10048–10060. doi:10.1016/j.tet.2005.08.018 |
| 13. | Ni, J.; Singh, S.; Wang, L.-X. Bioconjugate Chem. 2003, 14, 232–238. doi:10.1021/bc025617f |
| 14. | Sanki, A. K.; Mahal, L. K. Synlett 2006, 455–459. doi:10.1055/s-2006-926264 |
| 2. | Blixt, O.; Han, S.; Liao, L.; Zeng, Y.; Hoffmann, J.; Futakawa, S.; Paulson, J. C. J. Am. Chem. Soc. 2008, 130, 6680–6681. doi:10.1021/ja801052g |
| 3. | Huflejt, M. E.; Vuskovic, M.; Vasiliu, D.; Xu, H.; Obukhova, P.; Shilova, N.; Tuzikov, A.; Galanina, O.; Arun, B.; Lu, K.; Bovin, N. Mol. Immunol. 2009, 46, 3037–3049. doi:10.1016/j.molimm.2009.06.010 |
| 7. | Zhi, Z.; Laurent, N.; Powell, A. K.; Karamanska, R.; Fais, M.; Voglmeir, J.; Wright, A.; Blackburn, J. M.; Crocker, P. R.; Russell, D. A.; Flitsch, S. L.; Field, R. A.; Turnbull, J. E. ChemBioChem 2008, 9, 1568–1575. doi:10.1002/cbic.200700788 |
| 2. | Blixt, O.; Han, S.; Liao, L.; Zeng, Y.; Hoffmann, J.; Futakawa, S.; Paulson, J. C. J. Am. Chem. Soc. 2008, 130, 6680–6681. doi:10.1021/ja801052g |
| 3. | Huflejt, M. E.; Vuskovic, M.; Vasiliu, D.; Xu, H.; Obukhova, P.; Shilova, N.; Tuzikov, A.; Galanina, O.; Arun, B.; Lu, K.; Bovin, N. Mol. Immunol. 2009, 46, 3037–3049. doi:10.1016/j.molimm.2009.06.010 |
| 4. | Bejugam, M.; Flitsch, S. L. Org. Lett. 2004, 6, 4001–4004. doi:10.1021/ol048342n |
| 5. | Macmillan, D.; Daines, A. M.; Bayrhuber, M.; Flitsch, S. L. Org. Lett. 2002, 4, 1467–1470. doi:10.1021/ol025627w |
| 6. | Hartmann, M.; Horst, A. K.; Klemm, P.; Lindhorst, T. K. Chem. Commun. 2010, 46, 330–332. doi:10.1039/b922525k |
| 7. | Zhi, Z.; Laurent, N.; Powell, A. K.; Karamanska, R.; Fais, M.; Voglmeir, J.; Wright, A.; Blackburn, J. M.; Crocker, P. R.; Russell, D. A.; Flitsch, S. L.; Field, R. A.; Turnbull, J. E. ChemBioChem 2008, 9, 1568–1575. doi:10.1002/cbic.200700788 |
| 8. | Karamanska, R.; Clarke, J.; Blixt, O.; MacRae, J. I.; Zhang, J. Q.; Crocker, P. R.; Laurent, N.; Wright, A.; Flitsch, S. L.; Russell, D. A.; Field, R. A. Glycoconjugate J. 2008, 25, 69–74. doi:10.1007/s10719-007-9047-y |
| 9. | Laurent, N.; Voglmeir, J.; Wright, A.; Blackburn, J.; Pham, N. T.; Wong, S. C. C.; Gaskell, S. J.; Flitsch, S. L. ChemBioChem 2008, 9, 883–887. doi:10.1002/cbic.200700692 |
| 10. | Laurent, N.; Haddoub, R.; Flitsch, S. L. Trends Biotechnol. 2008, 26, 328–337. doi:10.1016/j.tibtech.2008.03.003 |
| 11. | Laurent, N.; Voglmeir, J.; Flitsch, S. L. Chem. Commun. 2008, 4400–4412. doi:10.1039/b806983m |
| 12. | Laurent, N.; Haddoub, R.; Voglmeir, J.; Wong, S. C. C.; Gaskell, S. J.; Flitsch, S. L. ChemBioChem 2008, 9, 2592–2596. doi:10.1002/cbic.200800481 |
| 18. | Noble, G. T.; Flitsch, S. L.; Liem, K. P.; Webb, S. J. Org. Biomol. Chem. 2009, 7, 5245–5254. doi:10.1039/b910976e |
| 17. | Chernyak, A. Y.; Sharma, G. V. M.; Kononov, L. O.; Krishna, P. R.; Levinsky, A. B.; Kochetkov, N. K.; Rama Rao, A. V. Carbohydr. Res. 1992, 223, 303–309. doi:10.1016/0008-6215(92)80029-Z |
© 2010 Šardzík et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)