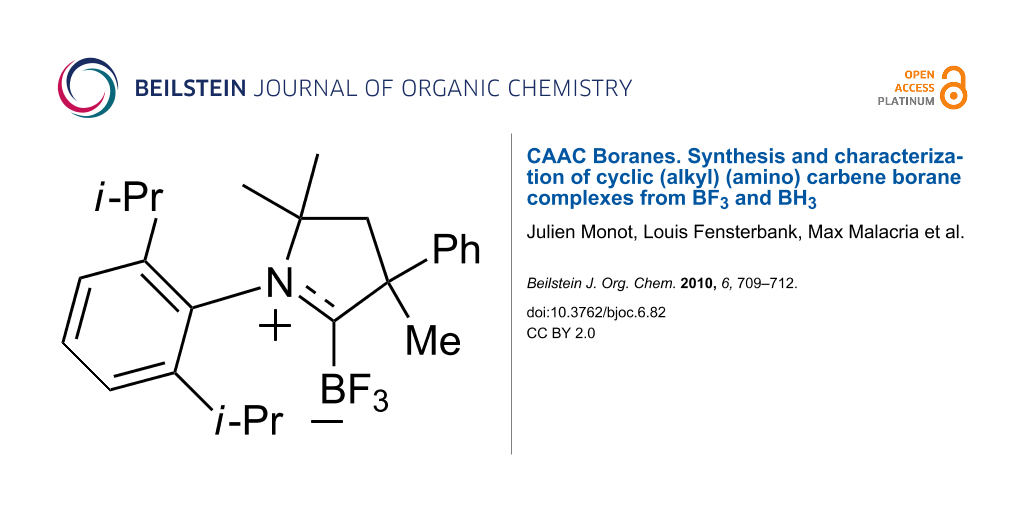
Abstract
In situ formation of two cyclic (alkyl) (amino) carbenes (CAACs) followed by addition of BF3•Et2O provided the first two examples of CAAC–BF3 complexes: 1-(2,6-diisopropylphenyl)-3,5,5-trimethyl-3-phenylpyrrolidin-2-ylidene trifluoroborane, and 2-(2,6-diisopropylphenyl)-3,3-dimethyl-2-azaspiro[4.5]decan-1-ylidene trifluoroborane. These CAAC–BF3 complexes are robust compounds that are stable to ambient laboratory conditions and silica gel chromatography. They were characterized by spectroscopy and X-ray crystallography. In contrast, a CAAC complex with borane (BH3) was readily formed in situ according to 1H and 11B NMR analysis, but did not survive the workup conditions. These results set the stage for further studies of the chemistry of CAAC boranes.

Graphical Abstract
Introduction
Lewis acid/Lewis base complexes of N-heterocyclic carbenes and boranes (NHC–boranes) are readily prepared from NHC’s and boranes by direct complexation [1-4]. Unlike many other classes of Lewis base complexes of boranes with neutral molecules (ethers, sulfides, etc.), NHC–borane complexes are highly stable in diverse environments. Complexes such as those shown in Figure 1 are white solids that can often be chromatographed if desired. Many such complexes resist decomplexation, oxidation, and both acidic and basic hydrolysis. Such NHC–boranes are beginning to be used as synthetic reagents, with applications in radical [5-9], ionic [10,11] and organometallic [12] reactions. They are also precursors for making higher complexes with unusual bonding patterns such as boron–boron double bonds [3], or for making unusual reactive intermediates on boron [6,7,13].
![[1860-5397-6-82-1]](/bjoc/content/figures/1860-5397-6-82-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative complexes of N-heterocyclic carbenes and boranes (NHC–boranes).
Figure 1: Representative complexes of N-heterocyclic carbenes and boranes (NHC–boranes).
Most of the first generation carbene-boranes have been made from N-heterocyclic carbenes in which the carbene carbon is stabilized by two donating nitrogen atoms (imidazolylidene, triazolylidene, etc) or other heteroatoms (phosphorous, silicon, oxygen) [14-18]. An exception is Bertrand’s BF3 complex of an acyclic (amino) (aryl) carbene in which the aryl group is 9-anthracenyl [19]. This complex, whose structure is shown in Figure 2, has also two cation stabilizing groups on the carbene carbon.
![[1860-5397-6-82-2]](/bjoc/content/figures/1860-5397-6-82-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Bertrand’s amino anthracenyl carbene trifluoroborane complex.
Figure 2: Bertrand’s amino anthracenyl carbene trifluoroborane complex.
Accordingly, it is interesting to study borane complexes of other types of carbenes. Recently, Bertrand introduced, cyclic (alkyl) (amino) carbenes, or CAACs, as a new class of carbene complex in which the central carbene carbon is stabilized by only one nitrogen atom [20-23]. Herein we report the synthesis and characterization (including X-ray structures) of two stable CAAC–BF3 complexes. We also show through spectroscopic studies that a representative CAAC–BH3 complex can be generated in solution.
Results and Discussion
CAAC trifluoroborane complexes 3a and 3b were readily prepared as summarized in Scheme 1. Deprotonation of the 3,4-dihydro-2H-pyrrolium hydrogen dichloride salt 1a with NaHMDS in THF at −78 °C, followed by warming to room temperature gave a pale yellow solution of the free carbene 2a [20]. A broad resonance at −312 ppm in the 13C NMR spectrum of this solution indicated that 2a had formed. Boron trifluoride etherate (BF3•Et2O) was then added at −78 °C and the resulting solution was stirred overnight. Rapid filtration through silica gel afforded the pure complex 3a (1-(2,6-diisopropylphenyl)-3,5,5-trimethyl-3-phenylpyrrolidin-2-ylidene trifluoroborane) as white crystals in 61% yield. CAAC borane 3b (2-(2,6-diisopropylphenyl)-3,3-dimethyl-2-azaspiro[4.5]decan-1-ylidene trifluoroborane) was made by a similar procedure starting from triflate salt 1b and was isolated in 64% yield.
![[1860-5397-6-82-i1]](/bjoc/content/inline/1860-5397-6-82-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of stable CAAC–BF3 complexes 3a and 3b and in situ generation of CAAC–BH3 complex 4a.
Scheme 1: Synthesis of stable CAAC–BF3 complexes 3a and 3b and in situ generation of CAAC–BH3 complex 4a.
CAAC boranes 3a and 3b were obtained as white crystals, and the structure of a single crystal of each was solved by X-ray diffraction. Two views of each structure are shown in Figure 3. Of special interest are the lengths of the Ccarbene–N bonds. At 1.294(3) Å for 3a and 1.293(3) Å for 3b, these bonds are significantly shorter than analogous bonds on other NHC–BF3 complexes where the carbene carbon has two nitrogen substituents (1.338(5)–1.355(5) Å) [24,25]. Instead, these bond lengths are closer to that of the BF3 complex of the amino-anthryl-carbene (1.301(2) Å) shown in Figure 2 [19]. Presumably, this is because there is more demand on the lone nitrogen of CAAC complex to stabilize the positive charge than when two nitrogen atoms are present. In contrast, the Ccarbene–B bond lengths of 3a (1.674(3) Å) and 3b (1.681(3) Å) are somewhat longer than those in other NHC–BF3 complexes (1.635(5)–1.668(6) Å) [24,25].
![[1860-5397-6-82-3]](/bjoc/content/figures/1860-5397-6-82-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-Ray crystal structures of CAAC–BF3 complexes 3a (top) and 3b (bottom).
Figure 3: X-Ray crystal structures of CAAC–BF3 complexes 3a (top) and 3b (bottom).
To conclude this preliminary study, we briefly attempted formation of the borane complex 4a by generation of carbene 2a as above, followed by the addition of BH3•THF. Indeed, complex 4a was directly formed in situ as evidenced by 11B NMR spectroscopy. The resonance for BH3•THF was absent and in its place was a new quartet at −30.0 ppm. Both the chemical shift and multiplicity of this resonance are consistent with the CAAC–borane structure 4a. The 1H NMR spectrum of the reaction mixture also showed the formation of 4a (see Supporting Information File 1).
The reaction mixture was allowed to stand for 15 h, during which time the 11B NMR spectrum was unchanged. This suggests that complex 4a is thermally stable. However, standard workup and evaporation as used for NHC–boranes and CAAC–BF3 complexes 3a and 3b did not provide 4a. Evidently, 4a is not as stable towards isolation as the BH3 complexes shown in Figure 1. Nonetheless, it should still be possible to generate this complex in situ and use it directly for onward reactions.
Conclusion
In summary, we have synthesized the first CAAC borane complexes. The complexes of trifluoroborane (BF3) are stable and were isolated as pure solids and fully characterized by spectroscopic analysis and X-ray crystallography. The borane (BH3) complex was characterized in situ by NMR spectroscopy, but did not survive workup and isolation. These results set the stage for further studies of the chemistry of CAAC boranes.
Supporting Information
| Supporting Information File 1: Procedures and characterization of the new complexes. | ||
| Format: PDF | Size: 316.8 KB | Download |
| Supporting Information File 2: Cif file of crystal structure of compound 3a. | ||
| Format: CIF | Size: 27.8 KB | Download |
| Supporting Information File 3: Cif file of crystal structure of compound 3b. | ||
| Format: CIF | Size: 12.9 KB | Download |
Acknowledgements
This work was supported by grants from the US National Science Foundation (CHE-0645998), UPMC, CNRS, and the French Agence Nationale de la Recherche (ANR, BLAN0309 Radicaux Verts, and 08-CEXC-011-01 Borane). We thank H. Rousselière and P. Herson (IPCM, Paris) for solving one of the crystal structures. We thank Dr. G. Mignani of Rhodia, Inc. for generously providing salt 1a.
References
-
Kuhn, N.; Henkel, G.; Kratz, T.; Kreutzberg, J.; Boese, R.; Maulitz, A. H. Chem. Ber. 1993, 126, 2041–2045. doi:10.1002/cber.19931260913
Return to citation in text: [1] -
Ramnial, T.; Jong, H.; McKenzie, I. D.; Jennings, M.; Clyburne, J. A. C. Chem. Commun. 2003, 1722–1723. doi:10.1039/b301416a
Return to citation in text: [1] -
Wang, Y.; Quillian, B.; Wei, P.; Wannere, C. S.; Xie, Y.; King, R. B.; Schaefer, H. F., III; v. R. Schleyer, P.; Robinson, G. H. J. Am. Chem. Soc. 2007, 129, 12412–12413. doi:10.1021/ja075932i
Return to citation in text: [1] [2] -
Yamaguchi, Y.; Kashiwabara, T.; Ogata, K.; Miura, Y.; Nakamura, Y.; Kobayashi, K.; Ito, T. Chem. Commun. 2004, 2160–2161. doi:10.1039/b405459h
Return to citation in text: [1] -
Ueng, S.-H.; Makhlouf Brahmi, M.; Derat, É.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D. P. J. Am. Chem. Soc. 2008, 130, 10082–10083. doi:10.1021/ja804150k
Return to citation in text: [1] -
Matsumoto, T.; Gabbaï, F. P. Organometallics 2009, 28, 4252–4253. doi:10.1021/om900476g
Return to citation in text: [1] [2] -
Ueng, S.-H.; Solovyev, A.; Yuan, X.; Geib, S. J.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Newcomb, M.; Walton, J. C.; Curran, D. P. J. Am. Chem. Soc. 2009, 131, 11256–11262. doi:10.1021/ja904103x
Return to citation in text: [1] [2] -
Walton, J. C.; Makhlouf Brahmi, M.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Chu, Q.; Ueng, S.-H.; Solovyev, A.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 2350–2358. doi:10.1021/ja909502q
Return to citation in text: [1] -
Tehfe, M.-A.; Makhlouf Brahmi, M.; Fouassier, J.-P.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E.; Lalevée, J. Macromolecules 2010, 43, 2261–2267. doi:10.1021/ma902492q
Return to citation in text: [1] -
Chu, Q.; Makhlouf Brahmi, M.; Solovyev, A.; Ueng, S.-H.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E. Chem.–Eur. J. 2009, 15, 12937–12940. doi:10.1002/chem.200902450
Return to citation in text: [1] -
Lindsay, D. M.; McArthur, D. Chem. Commun. 2010, 46, 2474–2476. doi:10.1039/c001466d
Return to citation in text: [1] -
Monot, J.; Makhlouf Brahmi, M.; Ueng, S.-H.; Robert, C.; Desage-El Murr, M.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E. Org. Lett. 2009, 11, 4914–4917. doi:10.1021/ol902012c
Return to citation in text: [1] -
Braunschweig, H.; Chiu, C.-W.; Radacki, K.; Kupfer, T. Angew. Chem., Int. Ed. 2010, 49, 2041–2044. doi:10.1002/anie.200906884
Return to citation in text: [1] -
Alcaraz, G.; Reed, R.; Baceiredo, A.; Bertrand, G. Chem. Commun. 1993, 1354–1355.
Return to citation in text: [1] -
Lambert, C.; Lopez-Solera, I.; Raithby, P. R. Organometallics 1996, 15, 452–455. doi:10.1021/om950427s
Return to citation in text: [1] -
Tamm, M.; Lügger, T.; Hahn, F. E. Organometallics 1996, 15, 1251–1256. doi:10.1021/om950773+
Return to citation in text: [1] -
Merceron, N.; Miqueu, K.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 2002, 124, 6806–6807. doi:10.1021/ja026556z
Return to citation in text: [1] -
Snead, D. R.; Ghiviriga, I.; Abboud, K. A.; Hong, S. Org. Lett. 2009, 11, 3274–3277. doi:10.1021/ol9013156
Return to citation in text: [1] -
Cattoën, X.; Gornitzka, H.; Bourissou, D.; Bertrand, G. J. Am. Chem. Soc. 2004, 126, 1342–1343. doi:10.1021/ja0396854
Return to citation in text: [1] [2] -
Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841
Return to citation in text: [1] [2] -
Lavallo, V.; Canac, Y.; DeHope, A.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 7236–7239. doi:10.1002/anie.200502566
Return to citation in text: [1] -
Jazzar, R.; Dewhurst, R. D.; Bourg, J.-B.; Donnadieu, B.; Canac, Y.; Bertrand, G. Angew. Chem., Int. Ed. 2007, 46, 2899–2902. doi:10.1002/anie.200605083
Return to citation in text: [1] -
Zeng, X.; Frey, G. D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131, 8690–8696. doi:10.1021/ja902051m
Return to citation in text: [1] -
Arduengo, A. J., III; Davidson, F.; Krafczyk, R.; Marshall, W. J.; Schmutzler, R. Monatsh. Chem. 2000, 131, 251–265. doi:10.1007/s007060070101
Return to citation in text: [1] [2] -
Nielsen, D. J.; Cavell, K. J.; Skelton, B. W.; White, A. H. Inorg. Chim. Acta 2003, 352, 143–150. doi:10.1016/S0020-1693(03)00143-9
Return to citation in text: [1] [2]
| 1. | Kuhn, N.; Henkel, G.; Kratz, T.; Kreutzberg, J.; Boese, R.; Maulitz, A. H. Chem. Ber. 1993, 126, 2041–2045. doi:10.1002/cber.19931260913 |
| 2. | Ramnial, T.; Jong, H.; McKenzie, I. D.; Jennings, M.; Clyburne, J. A. C. Chem. Commun. 2003, 1722–1723. doi:10.1039/b301416a |
| 3. | Wang, Y.; Quillian, B.; Wei, P.; Wannere, C. S.; Xie, Y.; King, R. B.; Schaefer, H. F., III; v. R. Schleyer, P.; Robinson, G. H. J. Am. Chem. Soc. 2007, 129, 12412–12413. doi:10.1021/ja075932i |
| 4. | Yamaguchi, Y.; Kashiwabara, T.; Ogata, K.; Miura, Y.; Nakamura, Y.; Kobayashi, K.; Ito, T. Chem. Commun. 2004, 2160–2161. doi:10.1039/b405459h |
| 3. | Wang, Y.; Quillian, B.; Wei, P.; Wannere, C. S.; Xie, Y.; King, R. B.; Schaefer, H. F., III; v. R. Schleyer, P.; Robinson, G. H. J. Am. Chem. Soc. 2007, 129, 12412–12413. doi:10.1021/ja075932i |
| 12. | Monot, J.; Makhlouf Brahmi, M.; Ueng, S.-H.; Robert, C.; Desage-El Murr, M.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E. Org. Lett. 2009, 11, 4914–4917. doi:10.1021/ol902012c |
| 10. | Chu, Q.; Makhlouf Brahmi, M.; Solovyev, A.; Ueng, S.-H.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E. Chem.–Eur. J. 2009, 15, 12937–12940. doi:10.1002/chem.200902450 |
| 11. | Lindsay, D. M.; McArthur, D. Chem. Commun. 2010, 46, 2474–2476. doi:10.1039/c001466d |
| 24. | Arduengo, A. J., III; Davidson, F.; Krafczyk, R.; Marshall, W. J.; Schmutzler, R. Monatsh. Chem. 2000, 131, 251–265. doi:10.1007/s007060070101 |
| 25. | Nielsen, D. J.; Cavell, K. J.; Skelton, B. W.; White, A. H. Inorg. Chim. Acta 2003, 352, 143–150. doi:10.1016/S0020-1693(03)00143-9 |
| 5. | Ueng, S.-H.; Makhlouf Brahmi, M.; Derat, É.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D. P. J. Am. Chem. Soc. 2008, 130, 10082–10083. doi:10.1021/ja804150k |
| 6. | Matsumoto, T.; Gabbaï, F. P. Organometallics 2009, 28, 4252–4253. doi:10.1021/om900476g |
| 7. | Ueng, S.-H.; Solovyev, A.; Yuan, X.; Geib, S. J.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Newcomb, M.; Walton, J. C.; Curran, D. P. J. Am. Chem. Soc. 2009, 131, 11256–11262. doi:10.1021/ja904103x |
| 8. | Walton, J. C.; Makhlouf Brahmi, M.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Chu, Q.; Ueng, S.-H.; Solovyev, A.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 2350–2358. doi:10.1021/ja909502q |
| 9. | Tehfe, M.-A.; Makhlouf Brahmi, M.; Fouassier, J.-P.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacôte, E.; Lalevée, J. Macromolecules 2010, 43, 2261–2267. doi:10.1021/ma902492q |
| 20. | Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841 |
| 21. | Lavallo, V.; Canac, Y.; DeHope, A.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 7236–7239. doi:10.1002/anie.200502566 |
| 22. | Jazzar, R.; Dewhurst, R. D.; Bourg, J.-B.; Donnadieu, B.; Canac, Y.; Bertrand, G. Angew. Chem., Int. Ed. 2007, 46, 2899–2902. doi:10.1002/anie.200605083 |
| 23. | Zeng, X.; Frey, G. D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131, 8690–8696. doi:10.1021/ja902051m |
| 24. | Arduengo, A. J., III; Davidson, F.; Krafczyk, R.; Marshall, W. J.; Schmutzler, R. Monatsh. Chem. 2000, 131, 251–265. doi:10.1007/s007060070101 |
| 25. | Nielsen, D. J.; Cavell, K. J.; Skelton, B. W.; White, A. H. Inorg. Chim. Acta 2003, 352, 143–150. doi:10.1016/S0020-1693(03)00143-9 |
| 19. | Cattoën, X.; Gornitzka, H.; Bourissou, D.; Bertrand, G. J. Am. Chem. Soc. 2004, 126, 1342–1343. doi:10.1021/ja0396854 |
| 19. | Cattoën, X.; Gornitzka, H.; Bourissou, D.; Bertrand, G. J. Am. Chem. Soc. 2004, 126, 1342–1343. doi:10.1021/ja0396854 |
| 14. | Alcaraz, G.; Reed, R.; Baceiredo, A.; Bertrand, G. Chem. Commun. 1993, 1354–1355. |
| 15. | Lambert, C.; Lopez-Solera, I.; Raithby, P. R. Organometallics 1996, 15, 452–455. doi:10.1021/om950427s |
| 16. | Tamm, M.; Lügger, T.; Hahn, F. E. Organometallics 1996, 15, 1251–1256. doi:10.1021/om950773+ |
| 17. | Merceron, N.; Miqueu, K.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 2002, 124, 6806–6807. doi:10.1021/ja026556z |
| 18. | Snead, D. R.; Ghiviriga, I.; Abboud, K. A.; Hong, S. Org. Lett. 2009, 11, 3274–3277. doi:10.1021/ol9013156 |
| 6. | Matsumoto, T.; Gabbaï, F. P. Organometallics 2009, 28, 4252–4253. doi:10.1021/om900476g |
| 7. | Ueng, S.-H.; Solovyev, A.; Yuan, X.; Geib, S. J.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Newcomb, M.; Walton, J. C.; Curran, D. P. J. Am. Chem. Soc. 2009, 131, 11256–11262. doi:10.1021/ja904103x |
| 13. | Braunschweig, H.; Chiu, C.-W.; Radacki, K.; Kupfer, T. Angew. Chem., Int. Ed. 2010, 49, 2041–2044. doi:10.1002/anie.200906884 |
| 20. | Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. doi:10.1002/anie.200501841 |
© 2010 Monot et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)