Abstract
The gold-catalyzed, seven-membered ring forming, intramolecular hydroamination of alkynic sulfonamides has been investigated. The protocol, with a semihollow-shaped triethynylphosphine as a ligand for gold, allowed the synthesis of a variety of azepine derivatives, which are difficult to access by other methods. Both alkynic sulfoamides with a flexible linear chain and the benzene-fused substrates underwent 7-exo-dig cyclization to afford the nitrogen-containing heterocyclic seven-membered rings, such as tetrahydroazepine and dihydrobenzazepine, in good yields.

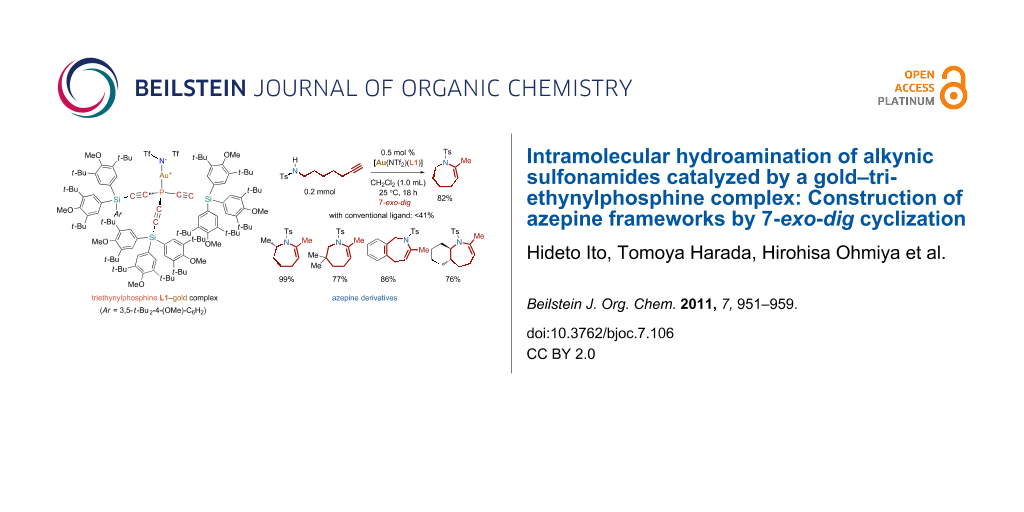
Graphical Abstract
Introduction
Nitrogen-containing heterocyclic seven-membered rings are found in many biologically active natural products and pharmaceuticals, such as (−)-tuberosutemonin (1) [1-6], related Stemona alkaloids [7], Cephalotaxus alkaloid (−)-cephalotaxine (2) [8-12], and SB-462795 (3) (Figure 1) [13-16]. Among a number of different approaches for the construction of N-heterocyclic compounds, metal-catalyzed intramolecular hydroamination of unactivated C–C multiple bonds is particularly straightforward and efficient [17,18]. Specifically, gold-catalyzed intramolecular hydroaminations of alkynes, alkenes and allenes show remarkable efficiency [19-28]. Unfortunately, however, the application of these methodologies to the synthesis of the N-heterocyclic seven-membered ring compounds is hampered by the low efficiency of seven-membered ring formations. Despite extensive studies on the gold-catalyzed intramolecular hydroamination of alkynes [19-51], seven-membered ring formation is rare and is limited to the cases where the substrate is preorganized for cyclization: The substrates must have geminal disubstitution or ring fusion within a linker chain connecting the attacking nitrogen atom and the alkyne moiety. It should be noted, however, that 7-“endo”-dig cyclizations of (o-alkynyl)phenylacetamides and a diynamide were achieved with gold and palladium complexes [39,46,52,53], and the zinc-catalyzed 7-exo-dig cyclization was reported specifically for a propargyl ether substrate [54].
![[1860-5397-7-106-1]](/bjoc/content/figures/1860-5397-7-106-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Azepine frameworks found in natural products and pharmaceuticals.
Figure 1: Azepine frameworks found in natural products and pharmaceuticals.
Previously, we reported that semihollow-shaped triethylnylphosphine L1 (Figure 2) exerted marked acceleration effects in the gold(I)-catalyzed Conia-ene reactions of acetylenic keto esters and enyne cycloisomerizations. The new catalytic system has expanded the scope of the reactions to six- and seven-membered ring formations, which had been difficult with the conventional catalytic systems [55]. Furthermore, we found that L1–gold(I) complex efficiently catalyzed the cyclization of internal alkyne substrates, which had also been difficult due to the steric repulsion between a nucleophilic center and a terminal substituent on the alkyne moiety [56]. We proposed that the cavity in the ligand forces the nucleophilic center closer to the gold-bound alkyne, resulting in the entropy-based rate enhancement. Recently, we further developed the gold(I)-catalyzed 7-exo-dig cyclization of acetylenic silyl enol ethers with L1 [57].
![[1860-5397-7-106-2]](/bjoc/content/figures/1860-5397-7-106-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Semihollow-shaped triethynylphosphine L1.
Figure 2: Semihollow-shaped triethynylphosphine L1.
In this context, we expected that the use of L1 as a ligand in the gold-catalyzed intramolecular hydroamination of alkynes would enable the construction of nitrogen-containing heterocyclic seven-membered rings, and we applied the triethynylphosphine–gold(I) catalytic systems to the synthesis of azepine derivatives through intramolecular hydroamination of alkynic sulfonamides. This article describes the results of the optimization of reaction conditions, exploration of substrate scope, and some mechanistic experiments.
Results and Discussion
Reaction conditions
The reaction conditions were optimized for the cyclization of N-(6-heptyn-1-yl)-p-toluenesulfonamide (4a) (Table 1). The triethynylphosphine–gold complex [Au(NTf2)(L1)] (0.5 mol %) catalyzed the cyclization of 4a (0.2 mmol) efficiently in CH2Cl2 (1.0 mL) at 25 °C (100% convn. of 4a) to afford 4,5,6,7-tetrahydroazepine derivative 5a with 18 h reaction time in 82% isolated yield (Table 1, entry 1). This reaction seemed to proceed through 7-exo-dig cyclization, but an exomethylene-type cyclic product 6a, which is a possible product of the 7-exo-dig cyclization [57], was not observed. The reaction under four-times-diluted conditions did not proceed to full conversion in the same reaction time (Table 1, entry 2). The reaction time was shortened to 9 h by heating at 80 °C, but this caused a slight decrease in the isolated yield of 5a (79%) (Table 1, entry 3).
Table 1: Optimization of reaction conditions for the gold-catalyzed intramolecular hydroamination of 4a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-106-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | Au cat. (mol %) | solvent (mL) | temp. (°C) | time (h) | convn. (%)a | yield orf 5 (%)a,b |
|---|---|---|---|---|---|---|
| 1 | [Au(NTf2)(L1)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 100 | 90 (82) |
| 2 | [Au(NTf2)(L1)] (0.5) | CH2Cl2 (4.0) | 25 | 18 | 87 | 85 |
| 3 | [Au(NTf2)(L1)] (0.5) | DCE (1.0) | 80 | 9 | 100 | 83 (79) |
| 4 | [Au(NTf2)(L1)] (0.5) | toluene (1.0) | 25 | 18 | 98 | 97 |
| 5 | [Au(NTf2)(L1)] (0.5) | THF (1.0) | 25 | 18 | 29 | 24 |
| 6 | [Au(NTf2)(L1)] (0.5) | MeCN (1.0) | 25 | 18 | 0 | n. d. |
| 7 | [Au(OTf)(L1)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 100 | 90 (80) |
| 8 | [Au(SbF6)(L1)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 14 | n. d. |
| 9 | [Au(BF4)(L1)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 7 | n. d. |
| 10 | [Au(NTf2)(PPh3)] (0.5) | CH2Cl2 (1.0) | 25 | 24 | 29 | 22 (26) |
| 11 | [Au(NTf2)(PPh3)] (5.0) | CH2Cl2 (1.0) | 25 | 18 | 100 | 66 (64) |
| 12 | [Au(NTf2){P(OPh)3}] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 54 | 39 |
| 13 | [Au(NTf2)(X-Phos)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 41 | 41 |
| 14 | [Au(NTf2)(IPr)] (0.5) | CH2Cl2 (1.0) | 25 | 18 | 53 | 41 |
aDetermined by 1H NMR. bIsolated yield in parentheses.
Among other solvents examined, toluene gave a result comparable with CH2Cl2 (Table 1, entries 1 and 4). On the other hand, polar and potentially coordinating solvents such as THF and MeCN were not effective in this reaction (Table 1, entries 5 and 6). The effect of the counter anion of the cationic gold complex is shown in Table 1, entries 1 and 7–9. While OTf− was as effective as NTf2− (Table 1, entry 7), SbF6− and BF4− inhibited the reaction completely (Table 1, entries 8 and 9).
The ligand effect is evaluated in Table 1, entries 1 and 10–14. The reaction proceeded slowly, even with a conventional phosphine ligand PPh3, such that the starting material was not fully consumed even after 24 h and the yield was as low as 26% (Table 1, entry 10). Increasing the catalyst loading of [Au(NTf2)(PPh3)] to 5.0 mol % caused full consumption of 4a, but the cyclization product was obtained only in 64% yield (Table 1, entry 11). The low yield relative to the conversion value is probably due to oligomerization and/or product decomposition as suggested by TLC and 1H NMR analysis of the crude mixture. A phosphite ligand P(OPh)3, which is comparable to triethynylphosphine L1 in electron-donor ability [58], was slightly more effective than PPh3, but was far less effective than L1 (Table 1, entry 12). Bulky and strongly electron-donating ligands such as X-Phos and IPr were as effective as the electron-deficient ligand P(OPh)3 (Table 1, entries 13 and 14). Accordingly, it is concluded that the acceleration effect by L1 is not due to an electronic effect rather a steric effect.
The time–conversion profiles shown in Figure 3 clearly indicate that the high catalytic efficiency with L1 is due to the improvement of the reaction kinetics and not the thermal stability of the catalyst. Although it was reported that Au(NTf2)(IPr) was somewhat unstable in the gold-catalyzed intermolecular hydroamination of alkyne under heating conditions [59], the deactivation of the gold catalyst with IPr and X-Phos was not significant under the present reaction conditions: The reactions with X-Phos and IPr ligands reached 100% and 84% conversions after 58 h, respectively (see Supporting Information File 1 for reaction profiles with longer reaction times).
![[1860-5397-7-106-3]](/bjoc/content/figures/1860-5397-7-106-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Time–conversion profiles for the gold-catalyzed cyclization of 4a with L1, X-Phos and IPr ligands.
Figure 3: Time–conversion profiles for the gold-catalyzed cyclization of 4a with L1, X-Phos and IPr ligands.
Effect of N-substituents
While alkynic o-nitrotoluenesulfonamide 4b did not react at all with 0.5 mol % of [Au(NTf2)(L1)] at room temperature (Table 2, entry 1), this substrate underwent 7-exo-dig cyclization upon increasing catalyst loading to 2.5 mol % and heating at 80 °C, giving N-nosylazepine derivative 5b in 76% isolated yield (Table 2, entry 2). N-Benzyloxycarbonyl (Cbz) and N-acetylazepine derivatives 5c and 5d were obtained in low yields through the cyclization of substrates 4c and 4d (Table 2, entries 3 and 4). On the other hand, the reactions of the substrates bearing N-tert-butoxycarbonyl (Boc) or N-p-methoxybenzyl (PMB) groups (4e,f) did not give the desired products at all (Table 2, entries 5 and 6). It seems that the reactivity of the substrates is affected by the balance between nucleophilicity of the nitrogen atom and acidity of the N–H bond as well as a steric factor.
Table 2: Effect of N-substitutents.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-106-i4.svg?max-width=637&scale=1.0)
|
|||||||
| entry | R | Au cat. (mol %) | solvent | temp. (°C) | time (h) | convn. (%)a | yield of 5 (%)a |
|---|---|---|---|---|---|---|---|
| 1 | Ns (4b) | 0.5 | CH2Cl2 | 25 | 18 | 0 | n. d. |
| 2 | Ns (4b) | 2.5 | DCE | 80 | 18 | 100 | 76b |
| 3 | Cbz (4c) | 2.5 | DCE | 80 | 24 | 97 | 33 |
| 4 | Ac (4d) | 2.5 | DCE | 80 | 24 | 63 | 18 |
| 5 | Boc (4e) | 2.5 | DCE | 80 | 24 | 17 | n. d. |
| 6 | PMB (4f) | 2.5 | DCE | 80 | 24 | 0 | n. d. |
aDetermined by 1H NMR. bIsolated yield.
Effect of substituents in acyclic linkers
Next, we explored the substrate scope by introducing one or two substituents in the acyclic linker chain of the alkynic N-tosylsulfonamide 4a (Table 3). The introduction of the substituents at the α or β positions relative to the alkyne moiety caused a significant decrease in the reactivity, but the cyclization of the substituted alkynic sulfonamide 4g–l proceeded smoothly, with 2.5–5.0 mol % catalyst loading at 80 °C, into full substrate conversion. Specifically, the substrate bearing an α-Me group (4g) derived from L-alanine was quantitatively converted into 2,7-dimethyltetrahydroazepine 5g with 2.5 mol % of [Au(NTf2)(L1)] (99% isolated yield, Table 3, entry 1). Although the substitution with bulkier iPr or Bn groups at the α-position in 4h and 4i resulted in even lower reactivities, the corresponding cyclization products 5h and 5i were obtained in high or good yields (Table 3, entries 2 and 3). The substrates (4j–l) with geminal disubstitution at the β-carbon also participated in the 7-exo-dig cyclization in good yields (Table 3, entries 4–6). Among the cyclization products (5a–l) described above, only the β,β-diphenyl-substituted sulfonamide 5k was contaminated with a small amount of exomethylene product 6k (Table 3, entry 5).
It should be noted that the geminal disubstitution in 4j–l caused a drastic decrease in the ease of the cyclization, which necessitated much more harsh reaction conditions (5 mol % Au, 80 °C, 4–12 h, Table 3, entries 4–6) than those for the reaction of the parent substrate 4a (0.5 mol % Au, 25 °C, 18 h, Table 1, entry 1). This means that the Thorpe–Ingold effect did not operate in the present case and that the substituents caused steric repulsion hindering the cyclization.
Table 3: Cyclization of alkynic sulfonamides with an acyclic linker.a
| entry | substrate | Au cat. (mol %) | time (h) | convn. (%)b | product | yield (%)c |
|---|---|---|---|---|---|---|
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-106-i5.svg?max-width=637&scale=0.9)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-106-i6.svg?max-width=637&scale=0.9)
|
|||||
| 1 | R = Me (4g) | 2.5 | 8 | 100 | R = Me (5g) | 99 |
| 2 | R = iPr (4h) | 5.0 | 12 | 100 | R = iPr (5h) | 88 |
| 3 | R = Bn (4i) | 5.0 | 12 | 100 | R = Bn (5i) | 71 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-106-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-106-i8.svg?max-width=637&scale=0.9)
|
|||||
| 4 | R = Me (4j) | 5.0 | 4 | 100 | R = Me (5j) | 77 |
| 5 | R = Ph (4k) | 5.0 | 4 | 100 | R = Ph (5k) | 69d |
| 6 | R = –(CH2)5– (4l) | 5.0 | 12 | 100 | R = –(CH2)5– (5l) | 66 |
aReaction conditions: 4, 0.1 mmol; [Au(NTf2)(L1)], 2.5 or 5 mol %; DCE, 1.0 mL; 80 °C.
bDetermined by 1H NMR.
cIsolated yield.
dMixture of 5k and 6k in 92:8 ratio. ![[Graphic 7]](/bjoc/content/inline/1860-5397-7-106-i9.svg?max-width=637&scale=0.945456)
Construction of bicyclic frameworks
Next, we applied the gold(I)–triethynylphosphine L1 complex to the construction of bicyclic frameworks such as benzazepine (Table 4). The cyclization of o-alkynyl benzylsulfonamide 4m proceeded with both [Au(NTf2)(L1)] and [Au(NTf2)(IPr)] to give a benzazepine derivative 5m (Table 4, entries 1 and 2). Although the starting material was fully consumed after 3 h or 6 h, using the respective catalysts, L1 was superior to IPr with respect to both reaction time and product yield. The reaction of N-tosylbenzamide 4n with L1 afforded the benzene-fused ε-caprolactam 6n within an exomethylene structure in 97% yield in an isomerically pure form (vide infra for discussion) (Table 4, entry 3). Sulfonamide 4o, with a cyclohexane-fused linker, also participated in the cyclization to form azabicylo[5.4.0]decene 5o in 76% yield along with a small amount of exomethylene isomer 6o (5o/6o 98:2, Table 4, entry 4).
Table 4: Cyclization of alkynic sulfonamide with a ring-fused linker.a
| entry | substrate | Au cat. (mol %) | time (h) | convn. (%)b | product | yield (%)c |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-106-i10.svg?max-width=637&scale=0.9)
4m |
[Au(NTf2)(L1)] (2.5) | 3 | 100 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-106-i11.svg?max-width=637&scale=0.9)
5m |
86 |
| 2 | 4m | [Au(NTf2)(IPr)] (2.5) | 6 | 100 | 5m | 58 |
| 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-106-i12.svg?max-width=637&scale=0.9)
4n |
[Au(NTf2)(L1)] (5.0) | 3 | 100 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-106-i13.svg?max-width=637&scale=0.9)
6n |
97 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-106-i14.svg?max-width=637&scale=0.9)
4o |
[Au(NTf2)(L1)] (2.5) | 17 | 100 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-106-i15.svg?max-width=637&scale=0.9)
5o |
76d |
aReaction conditions: 4, 0.1 mmol; DCE, 1.0 mL; 80 °C.
bDetermined by 1H NMR.
cIsolated yield.
dMixture of 5o and 6o in 98:2 ratio. ![[Graphic 14]](/bjoc/content/inline/1860-5397-7-106-i16.svg?max-width=637&scale=0.945456)
Effect of ring sizes
We also evaluated the triethynylphosphine L1, X-Phos, and IPr for an acceleration effect in the six-membered, ring forming, gold-catalyzed hydroamination of N-(5-hexyn-1-yl)-p-toluenesulfonamide 7. As expected from entropy considerations, the six-membered ring formations of 7 with these ligands were generally much faster than the seven-membered ring formations of 4: The reaction with 0.5 mol % catalyst loading at room temperature completed within 1 h irrespective of the ligand used. When the catalyst loading was reduced to 0.1 mol %, however, the superiority of L1 to X-Phos and IPr became significant, as shown in Table 5. The reaction with L1 at room temperature afforded the six-membered ring product 8 in 91% isolated yield after 2 h (Table 1, entry 1). On the other hand, the reaction with X-Phos did not reach full conversion (76% convn.) even after 12 h and gave 8 in only 70% yield (Table 5, entry 2). The use of IPr ligand resulted in even lower conversion (68%) and isolated yield (58%) (Table 5, entry 3).
Table 5: 6-exo-dig cyclization of sulfonamide 7.
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-106-i17.svg?max-width=637&scale=1.0)
|
||||
| entry | Ligand |
time
(h) |
convn.
(%)a |
yield of 8
(%)a,b |
|---|---|---|---|---|
| 1 | L1 | 2 | 100 | 100 (91) |
| 2 | X-Phos | 12 | 76 | 76 (70) |
| 3 | IPr | 12 | 68 | 67 (58) |
aDetermined by 1H NMR. bIsolated yield in the parentheses.
The triethynylphosphine ligand L1 was also evaluated for the eight-membered ring formation of sulfonamide 9, which is much more challenging than the seven-membered ring formation of 4. The reaction required 5 mol % catalyst loading under heating conditions (80 °C) in 1,2-dichloroethane for a reasonable conversion rate to afforded an eight-membered ring azocine derivative 10 in 15% isolated yield (Scheme 1). It should be noted that the reaction produced significant amounts of unidentified oligomeric side products.
![[1860-5397-7-106-i1]](/bjoc/content/inline/1860-5397-7-106-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 8-exo-dig cyclization of sulfonamide 9.
Scheme 1: 8-exo-dig cyclization of sulfonamide 9.
Alkene isomerization
We carried out alkene isomerization experiments to clarify how tetrahydroazepines 5 formed via the 7-exo-dig cyclization of 4. One possible reaction pathway is the alkene isomerization of an exomethylene product 6. To test this possibility, we synthesized 6a through another route (see Supporting Information File 1) and subjected it to the standard reaction conditions of the gold–triethynylphosphine-catalyzed cyclization of alkynic sulfonamide with or without N-tosyl aniline 7 as an external proton source (Scheme 2). Although 6a was indeed isomerized into 5a to some extent in both cases, the main reaction was decomposition to give complex mixtures. The exomethylene substrate 6a appeared to be unstable at room temperature even in the absence of the gold-catalyst.
![[1860-5397-7-106-i2]](/bjoc/content/inline/1860-5397-7-106-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Isomerization experiments of 6a.
Scheme 2: Isomerization experiments of 6a.
According to these results, the formation of exomethylene compound 6 and subsequent alkene isomerisation should not be a major pathway to 5. Instead, a possible reaction pathway from 4a to 5a is illustrated in Figure 4. First, the cationic gold center coordinates with 4a to form the gold–alkyne complex A. Intramolecular nucleophilic attack of the nitrogen atom affords the 7-exo-dig cyclization product B with an exocyclic C–C double bond. The protonated N-sulfonylenamide B tautomerizes to iminium ion C through 1,3-proton shift, or through an alternative pathway via a gold(I)–carben intermediate (D). Then, re-tautomerization affords the protonated N-sulfonylenamide E with an endocyclic C–C double bond. Finally, protodemetalation of E give the N-sulfonylenamide 5a, which is thermodynamically more stable than 6a.
![[1860-5397-7-106-4]](/bjoc/content/figures/1860-5397-7-106-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Possible pathway for the gold-catalyzed conversion of 4a into 5a.
Figure 4: Possible pathway for the gold-catalyzed conversion of 4a into 5a.
It should be noted that the reaction of the N-tosylbenzamide 4n afforded exceptionally the exomethylene isomer 6n. One conceivable reason is that the alkene isomerisation was prevented due to a ring strain in the seven-membered ring of 5n, of which six out of seven atoms are sp2-hybridized.
Conclusion
We demonstrated that the 7-exo-dig intramolecular hydroamination of ω-alkynic N-alkyl-N-sulfonamides is efficiently catalyzed by a gold(I) complex coordinated with the semihollow-shaped triethynylphopshine ligand L1, and that the cyclization protocol provides a new efficient route to N-containing seven-membered ring compounds. The protocol is applicable to the reaction of alkynic sulfonamides with an acyclic or ring-fused linker chain with various substitution patterns. Evaluation of the ligand effect in the gold catalysis with different ligands and substrates strongly suggested that the rate enhancement by the triethynylphosphine would be due to a steric factor which enforces a nucleophilic center close to a gold-activated alkyne moiety.
Experimental
Preparation of [Au(NTf2)(L1)]
[AuCl(L1)] (1 equiv) was placed in an open vial and was dissolved in CH2Cl2 (ca. 0.1 M). AgNTf2 (>1.5 equiv) was added, and the mixture was stirred at 25 °C for 10 min. The resulting white suspension was filtered through celite into a screw vial. The resulting colorless solution was first concentrated with a stream of Ar gas and then dried in vacuo to give [Au(NTf2)(L1)] as a white solid. (See also [55].)
General procedure for gold-catalyzed intramolecular hydroamination of alkynic sulfonamide 4a
[Au(NTf2)(L1)] (2.6 mg, 1.0 μmol, 0.5 mol %) and a magnetic stirring bar were placed in an open vial. Separately, the alkynyl sulfonamide 4a (55 mg, 0.20 mmol) was weighted into a micro tube. The tubes were placed in a glove box. The gold complex and 4a were dissolved in degassed dry CH2Cl2 (0.25 mL), in their respective tubes. The solution of 4a was transferred to the solution of the catalyst with a syringe. The remaining solutions in the micro tube and the syringe were washed with CH2Cl2 (2 × 0.25 mL) and, the washings were added to the reaction mixture. The tube was sealed with a cap equipped with a Teflon-coated silicon rubber septum. The tube was taken from the glove box, and was placed in a water bath (25 °C). After the reaction was complete (as monitored by TLC), the reaction mixture was passed through a pad of silica gel and was concentrated to dryness. Purification by flash chromatography on silica gel gave the cyclization product 5a (45 mg, 82%) as a white solid; mp 65.8–66.1 °C; 1H NMR (300 MHz, CDCl3) δ 1.32–1.53 (m, 6H), 1.94 (t, J = 2.7 Hz, 1H), 2.14 (td, J = 6.9, 2.7 Hz, 2H), 2.44 (s, 3H), 2.95 (q, J = 6.9 Hz, 2H), 4.34 (br s, 1H), 7.31 (d, J = 8.1 Hz, 2H), 7.75 (d, J = 8.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 17.94, 21.29, 25.34, 27.59, 28.75, 42.80, 68.35, 84.05, 127.06, 129.90, 136.85, 143.34; Anal. calcd for C14H19NO2S: C, 63.36; H, 7.22; N, 5.28; found: C, 63.29; H, 7.16; N, 5.21.
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR spectra for 4a–o and 5a, b, g–m, o, 6a, n, 7, 8, 9, 10. | ||
| Format: PDF | Size: 261.6 KB | Download |
References
-
Uyeo, S.; Irie, H.; Harada, H. Chem. Pharm. Bull. 1967, 15, 768–770.
Return to citation in text: [1] -
Harada, H.; Irie, H.; Masaki, N.; Osaki, K.; Uyeo, S. Chem. Commun. (London) 1967, 460–462. doi:10.1039/c19670000460
Return to citation in text: [1] -
Götz, M.; Bögri, T.; Gray, A. H.; Strunz, G. M. Tetrahedron 1968, 24, 2631–2643. doi:10.1016/S0040-4020(01)82538-2
Return to citation in text: [1] -
Lin, W.-H.; Ye, Y.; Xu, R.-S. J. Nat. Prod. 1992, 55, 571–576. doi:10.1021/np50083a003
Return to citation in text: [1] -
Pilli, R. A.; Ferreira de Oliveira, M. C. Nat. Prod. Rep. 2000, 17, 117–127. doi:10.1039/a902437i
Return to citation in text: [1] -
Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t
Return to citation in text: [1] -
Alibés, R.; Figueredo, M. Eur. J. Org. Chem. 2009, 2009, 2421–2435. doi:10.1002/ejoc.200900037
Return to citation in text: [1] -
Paudler, W. W.; Kerley, G. I.; McKay, J. J. Org. Chem. 1963, 28, 2194–2197. doi:10.1021/jo01044a010
Return to citation in text: [1] -
Weinreb, S. M.; Semmelhack, M. F. Acc. Chem. Res. 1975, 8, 158–164. doi:10.1021/ar50089a003
Return to citation in text: [1] -
Huang, L.; Xue, Z. Cephalotaxus alkaloids. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Academic Press: New York, 1984; Vol. 23, pp 157–226. doi:10.1016/S0099-9598(08)60071-1
Return to citation in text: [1] -
Jalil Miah, M. A.; Hudlicky, T.; Reed, J. W. Cephalotaxus Alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: San Diego, 1998; Vol. 51, pp 199–269. doi:10.1016/S0099-9598(08)60006-1
Return to citation in text: [1] -
Isono, N.; Mori, M. J. Org. Chem. 1995, 60, 115–119. doi:10.1021/jo00106a023
Return to citation in text: [1] -
Kumar, S.; Dare, L.; Vasko-Moser, J. A.; James, I. E.; Blake, S. M.; Rickard, D. J.; Hwang, S.-M.; Tomaszek, T.; Yamashita, D. S.; Marquis, R. W.; Oh, H.; Jeong, J. U.; Veber, D. F.; Gowen, M.; Lark, M. W.; Stroup, G. Bone 2007, 40, 122–131. doi:10.1016/j.bone.2006.07.015
Return to citation in text: [1] -
Yamashita, D. S.; Marquis, R. W.; Xie, R.; Nidamarthy, S. D.; Oh, H.-J.; Jeong, J. U.; Erhard, K. F.; Ward, K. W.; Roethke, T. J.; Smith, B. R.; Cheng, H.-Y.; Geng, X.; Lin, F.; Offen, P. H.; Wang, B.; Nevins, N.; Head, M. S.; Haltiwanger, R. C.; Narducci Sarjeant, A. A.; Liable-Sands, L. M.; Zhao, B.; Smith, W. W.; Janson, C. A.; Gao, E.; Tomaszek, T.; McQueney, M.; James, I. E.; Gress, C. J.; Zembryki, D. L.; Lark, M. W.; Veber, D. F. J. Med. Chem. 2006, 49, 1597–1612. doi:10.1021/jm050915u
Return to citation in text: [1] -
Wang, H.; Matsuhashi, H.; Doan, B. D.; Goodman, S. N.; Ouyang, X.; Clark, W. M., Jr. Tetrahedron 2009, 65, 6291–6303. doi:10.1016/j.tet.2009.06.022
Return to citation in text: [1] -
Goodman, S. N.; Dai, Q.; Wang, J.; Clark, W. M., Jr. Org. Process Res. Dev. 2011, 15, 123–130. doi:10.1021/op100266s
Return to citation in text: [1] -
Müller, T. E.; Beller, M. Chem. Rev. 1998, 98, 675–704. doi:10.1021/cr960433d
Return to citation in text: [1] -
Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] [2] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] [2] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] [2] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] [2] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] [2] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] [2] -
Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k
Return to citation in text: [1] [2] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] [2] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u
Return to citation in text: [1] [2] -
Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088
Return to citation in text: [1] [2] -
Shu, X.-Z.; Liu, X.-Y.; Xiao, H.-Q.; Ji, K.-G.; Guo, L.-N.; Liang, Y.-M. Adv. Synth. Catal. 2008, 350, 243–248. doi:10.1002/adsc.200700452
Return to citation in text: [1] -
Enomoto, T.; Obika, S.; Yasui, Y.; Takemoto, Y. Synlett 2008, 1647–1650. doi:10.1055/s-2008-1077879
Return to citation in text: [1] -
Ye, D.; Wang, J.; Zhang, X.; Zhou, Y.; Ding, X.; Feng, E.; Sun, H.; Liu, G.; Jiang, H.; Liu, H. Green Chem. 2009, 11, 1201–1208. doi:10.1039/b904044g
Return to citation in text: [1] -
Peng, H. M.; Zhao, J.; Li, X. Adv. Synth. Catal. 2009, 351, 1371–1377. doi:10.1002/adsc.200800735
Return to citation in text: [1] -
Ye, D.; Zhang, X.; Zhou, Y.; Zhang, D.; Zhang, L.; Wang, H.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2009, 351, 2770–2778. doi:10.1002/adsc.200900505
Return to citation in text: [1] -
Liu, X.-Y.; Che, C.-M. Org. Lett. 2009, 11, 4204–4207. doi:10.1021/ol901443b
Return to citation in text: [1] -
Yamane, Y.; Liu, X.; Hamasaki, A.; Ishida, T.; Haruta, M.; Yokoyama, T.; Tokunaga, M. Org. Lett. 2009, 11, 5162–5165. doi:10.1021/ol902061j
Return to citation in text: [1] -
Zhou, Y.; Feng, E.; Liu, G.; Ye, D.; Li, J.; Jiang, H.; Liu, H. J. Org. Chem. 2009, 74, 7344–7348. doi:10.1021/jo901418m
Return to citation in text: [1] -
Enomoto, T.; Girard, A.-L.; Yasui, Y.; Takemoto, Y. J. Org. Chem. 2009, 74, 9158–9164. doi:10.1021/jo901906b
Return to citation in text: [1] -
Han, Z.-Y.; Xiao, H.; Chen, X.-H.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 9182–9183. doi:10.1021/ja903547q
Return to citation in text: [1] -
Wilckens, K.; Uhlemann, M.; Czekelius, C. Chem.–Eur. J. 2009, 15, 13323–13326. doi:10.1002/chem.200901702
Return to citation in text: [1] [2] -
Hirano, K.; Inaba, Y.; Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Adv. Synth. Catal. 2010, 352, 368–372. doi:10.1002/adsc.200900880
Return to citation in text: [1] -
Zeng, X.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 942–945. doi:10.1002/anie.200905341
Return to citation in text: [1] -
Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. Adv. Synth. Catal. 2010, 352, 971–975. doi:10.1002/adsc.201000011
Return to citation in text: [1] -
Patil, N. T.; Singh, V.; Konala, A.; Mutyala, A. K. Tetrahedron Lett. 2010, 51, 1493–1496. doi:10.1016/j.tetlet.2010.01.036
Return to citation in text: [1] -
Gimeno, A.; Medio-Simón, M.; Ramírez de Arellano, C.; Asensio, G.; Cuenca, A. B. Org. Lett. 2010, 12, 1900–1903. doi:10.1021/ol100595s
Return to citation in text: [1] -
Wang, C.; Han, Z.-Y.; Luo, H.-W.; Gong, L.-Z. Org. Lett. 2010, 12, 2266–2269. doi:10.1021/ol1006086
Return to citation in text: [1] -
Zhang, L.; Ye, D.; Zhou, Y.; Liu, G.; Feng, E.; Jiang, H.; Liu, H. J. Org. Chem. 2010, 75, 3671–3677. doi:10.1021/jo100378u
Return to citation in text: [1] [2] -
Kothandaraman, P.; Rao, W.; Foo, S. J.; Chan, P. W. H. Angew. Chem., Int. Ed. 2010, 49, 4619–4623. doi:10.1002/anie.201000341
Return to citation in text: [1] -
Demir, A. S.; Emrullahoğlu, M.; Buran, K. Chem. Commun. 2010, 46, 8032–8034. doi:10.1039/c0cc02357d
Return to citation in text: [1] -
Monge, D.; Jensen, K. L.; Franke, P. T.; Lykke, L.; Jørgensen, K. A. Chem.–Eur. J. 2010, 16, 9478–9484. doi:10.1002/chem.201001123
Return to citation in text: [1] -
Arcadi, A.; Abbiati, G.; Rossi, E. J. Organomet. Chem. 2011, 696, 87–98. doi:10.1016/j.jorganchem.2010.08.011
Return to citation in text: [1] -
Kitahara, H.; Sakurai, H. J. Organomet. Chem. 2011, 696, 442–449. doi:10.1016/j.jorganchem.2010.08.038
Return to citation in text: [1] -
Zulys, A.; Dochnahl, M.; Hollmann, D.; Löhnwitz, K.; Herrmann, J.-S.; Roesky, P. W.; Blechert, S. Angew. Chem., Int. Ed. 2005, 44, 7794–7798. doi:10.1002/anie.200502006
Return to citation in text: [1] -
Yu, Y.; Stephenson, G. A.; Mitchell, D. Tetrahedron Lett. 2006, 47, 3811–3814. doi:10.1016/j.tetlet.2006.03.198
Return to citation in text: [1] -
Liu, J.; Shen, M.; Zhang, Y.; Li, G.; Khodabocus, A.; Rodriguez, S.; Qu, B.; Farina, V.; Senanayake, C. H.; Lu, B. Z. Org. Lett. 2006, 8, 3573–3575. doi:10.1021/ol061440j
Return to citation in text: [1] -
Ochida, A.; Ito, H.; Sawamura, M. J. Am. Chem. Soc. 2006, 128, 16486–16487. doi:10.1021/ja066800c
Return to citation in text: [1] [2] -
Ito, H.; Makida, Y.; Ochida, A.; Ohmiya, H.; Sawamura, M. Org. Lett. 2008, 10, 5051–5054. doi:10.1021/ol802079r
Return to citation in text: [1] -
Ito, H.; Ohmiya, H.; Sawamura, M. Org. Lett. 2010, 12, 4380–4383. doi:10.1021/ol101860j
Return to citation in text: [1] [2] -
Ochida, A.; Sawamura, M. Chem.–Asian J. 2007, 2, 609–618. doi:10.1002/asia.200700006
Return to citation in text: [1] -
Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093
Return to citation in text: [1]
| 1. | Uyeo, S.; Irie, H.; Harada, H. Chem. Pharm. Bull. 1967, 15, 768–770. |
| 2. | Harada, H.; Irie, H.; Masaki, N.; Osaki, K.; Uyeo, S. Chem. Commun. (London) 1967, 460–462. doi:10.1039/c19670000460 |
| 3. | Götz, M.; Bögri, T.; Gray, A. H.; Strunz, G. M. Tetrahedron 1968, 24, 2631–2643. doi:10.1016/S0040-4020(01)82538-2 |
| 4. | Lin, W.-H.; Ye, Y.; Xu, R.-S. J. Nat. Prod. 1992, 55, 571–576. doi:10.1021/np50083a003 |
| 5. | Pilli, R. A.; Ferreira de Oliveira, M. C. Nat. Prod. Rep. 2000, 17, 117–127. doi:10.1039/a902437i |
| 6. | Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t |
| 17. | Müller, T. E.; Beller, M. Chem. Rev. 1998, 98, 675–704. doi:10.1021/cr960433d |
| 18. | Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788 |
| 59. | Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093 |
| 13. | Kumar, S.; Dare, L.; Vasko-Moser, J. A.; James, I. E.; Blake, S. M.; Rickard, D. J.; Hwang, S.-M.; Tomaszek, T.; Yamashita, D. S.; Marquis, R. W.; Oh, H.; Jeong, J. U.; Veber, D. F.; Gowen, M.; Lark, M. W.; Stroup, G. Bone 2007, 40, 122–131. doi:10.1016/j.bone.2006.07.015 |
| 14. | Yamashita, D. S.; Marquis, R. W.; Xie, R.; Nidamarthy, S. D.; Oh, H.-J.; Jeong, J. U.; Erhard, K. F.; Ward, K. W.; Roethke, T. J.; Smith, B. R.; Cheng, H.-Y.; Geng, X.; Lin, F.; Offen, P. H.; Wang, B.; Nevins, N.; Head, M. S.; Haltiwanger, R. C.; Narducci Sarjeant, A. A.; Liable-Sands, L. M.; Zhao, B.; Smith, W. W.; Janson, C. A.; Gao, E.; Tomaszek, T.; McQueney, M.; James, I. E.; Gress, C. J.; Zembryki, D. L.; Lark, M. W.; Veber, D. F. J. Med. Chem. 2006, 49, 1597–1612. doi:10.1021/jm050915u |
| 15. | Wang, H.; Matsuhashi, H.; Doan, B. D.; Goodman, S. N.; Ouyang, X.; Clark, W. M., Jr. Tetrahedron 2009, 65, 6291–6303. doi:10.1016/j.tet.2009.06.022 |
| 16. | Goodman, S. N.; Dai, Q.; Wang, J.; Clark, W. M., Jr. Org. Process Res. Dev. 2011, 15, 123–130. doi:10.1021/op100266s |
| 55. | Ochida, A.; Ito, H.; Sawamura, M. J. Am. Chem. Soc. 2006, 128, 16486–16487. doi:10.1021/ja066800c |
| 8. | Paudler, W. W.; Kerley, G. I.; McKay, J. J. Org. Chem. 1963, 28, 2194–2197. doi:10.1021/jo01044a010 |
| 9. | Weinreb, S. M.; Semmelhack, M. F. Acc. Chem. Res. 1975, 8, 158–164. doi:10.1021/ar50089a003 |
| 10. | Huang, L.; Xue, Z. Cephalotaxus alkaloids. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Academic Press: New York, 1984; Vol. 23, pp 157–226. doi:10.1016/S0099-9598(08)60071-1 |
| 11. | Jalil Miah, M. A.; Hudlicky, T.; Reed, J. W. Cephalotaxus Alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: San Diego, 1998; Vol. 51, pp 199–269. doi:10.1016/S0099-9598(08)60006-1 |
| 12. | Isono, N.; Mori, M. J. Org. Chem. 1995, 60, 115–119. doi:10.1021/jo00106a023 |
| 57. | Ito, H.; Ohmiya, H.; Sawamura, M. Org. Lett. 2010, 12, 4380–4383. doi:10.1021/ol101860j |
| 7. | Alibés, R.; Figueredo, M. Eur. J. Org. Chem. 2009, 2009, 2421–2435. doi:10.1002/ejoc.200900037 |
| 58. | Ochida, A.; Sawamura, M. Chem.–Asian J. 2007, 2, 609–618. doi:10.1002/asia.200700006 |
| 54. | Liu, J.; Shen, M.; Zhang, Y.; Li, G.; Khodabocus, A.; Rodriguez, S.; Qu, B.; Farina, V.; Senanayake, C. H.; Lu, B. Z. Org. Lett. 2006, 8, 3573–3575. doi:10.1021/ol061440j |
| 56. | Ito, H.; Makida, Y.; Ochida, A.; Ohmiya, H.; Sawamura, M. Org. Lett. 2008, 10, 5051–5054. doi:10.1021/ol802079r |
| 39. | Wilckens, K.; Uhlemann, M.; Czekelius, C. Chem.–Eur. J. 2009, 15, 13323–13326. doi:10.1002/chem.200901702 |
| 46. | Zhang, L.; Ye, D.; Zhou, Y.; Liu, G.; Feng, E.; Jiang, H.; Liu, H. J. Org. Chem. 2010, 75, 3671–3677. doi:10.1021/jo100378u |
| 52. | Zulys, A.; Dochnahl, M.; Hollmann, D.; Löhnwitz, K.; Herrmann, J.-S.; Roesky, P. W.; Blechert, S. Angew. Chem., Int. Ed. 2005, 44, 7794–7798. doi:10.1002/anie.200502006 |
| 53. | Yu, Y.; Stephenson, G. A.; Mitchell, D. Tetrahedron Lett. 2006, 47, 3811–3814. doi:10.1016/j.tetlet.2006.03.198 |
| 57. | Ito, H.; Ohmiya, H.; Sawamura, M. Org. Lett. 2010, 12, 4380–4383. doi:10.1021/ol101860j |
| 19. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 20. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 21. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 22. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 23. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 24. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 25. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k |
| 26. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 27. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 28. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088 |
| 29. | Shu, X.-Z.; Liu, X.-Y.; Xiao, H.-Q.; Ji, K.-G.; Guo, L.-N.; Liang, Y.-M. Adv. Synth. Catal. 2008, 350, 243–248. doi:10.1002/adsc.200700452 |
| 30. | Enomoto, T.; Obika, S.; Yasui, Y.; Takemoto, Y. Synlett 2008, 1647–1650. doi:10.1055/s-2008-1077879 |
| 31. | Ye, D.; Wang, J.; Zhang, X.; Zhou, Y.; Ding, X.; Feng, E.; Sun, H.; Liu, G.; Jiang, H.; Liu, H. Green Chem. 2009, 11, 1201–1208. doi:10.1039/b904044g |
| 32. | Peng, H. M.; Zhao, J.; Li, X. Adv. Synth. Catal. 2009, 351, 1371–1377. doi:10.1002/adsc.200800735 |
| 33. | Ye, D.; Zhang, X.; Zhou, Y.; Zhang, D.; Zhang, L.; Wang, H.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2009, 351, 2770–2778. doi:10.1002/adsc.200900505 |
| 34. | Liu, X.-Y.; Che, C.-M. Org. Lett. 2009, 11, 4204–4207. doi:10.1021/ol901443b |
| 35. | Yamane, Y.; Liu, X.; Hamasaki, A.; Ishida, T.; Haruta, M.; Yokoyama, T.; Tokunaga, M. Org. Lett. 2009, 11, 5162–5165. doi:10.1021/ol902061j |
| 36. | Zhou, Y.; Feng, E.; Liu, G.; Ye, D.; Li, J.; Jiang, H.; Liu, H. J. Org. Chem. 2009, 74, 7344–7348. doi:10.1021/jo901418m |
| 37. | Enomoto, T.; Girard, A.-L.; Yasui, Y.; Takemoto, Y. J. Org. Chem. 2009, 74, 9158–9164. doi:10.1021/jo901906b |
| 38. | Han, Z.-Y.; Xiao, H.; Chen, X.-H.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 9182–9183. doi:10.1021/ja903547q |
| 39. | Wilckens, K.; Uhlemann, M.; Czekelius, C. Chem.–Eur. J. 2009, 15, 13323–13326. doi:10.1002/chem.200901702 |
| 40. | Hirano, K.; Inaba, Y.; Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Adv. Synth. Catal. 2010, 352, 368–372. doi:10.1002/adsc.200900880 |
| 41. | Zeng, X.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 942–945. doi:10.1002/anie.200905341 |
| 42. | Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. Adv. Synth. Catal. 2010, 352, 971–975. doi:10.1002/adsc.201000011 |
| 43. | Patil, N. T.; Singh, V.; Konala, A.; Mutyala, A. K. Tetrahedron Lett. 2010, 51, 1493–1496. doi:10.1016/j.tetlet.2010.01.036 |
| 44. | Gimeno, A.; Medio-Simón, M.; Ramírez de Arellano, C.; Asensio, G.; Cuenca, A. B. Org. Lett. 2010, 12, 1900–1903. doi:10.1021/ol100595s |
| 45. | Wang, C.; Han, Z.-Y.; Luo, H.-W.; Gong, L.-Z. Org. Lett. 2010, 12, 2266–2269. doi:10.1021/ol1006086 |
| 46. | Zhang, L.; Ye, D.; Zhou, Y.; Liu, G.; Feng, E.; Jiang, H.; Liu, H. J. Org. Chem. 2010, 75, 3671–3677. doi:10.1021/jo100378u |
| 47. | Kothandaraman, P.; Rao, W.; Foo, S. J.; Chan, P. W. H. Angew. Chem., Int. Ed. 2010, 49, 4619–4623. doi:10.1002/anie.201000341 |
| 48. | Demir, A. S.; Emrullahoğlu, M.; Buran, K. Chem. Commun. 2010, 46, 8032–8034. doi:10.1039/c0cc02357d |
| 49. | Monge, D.; Jensen, K. L.; Franke, P. T.; Lykke, L.; Jørgensen, K. A. Chem.–Eur. J. 2010, 16, 9478–9484. doi:10.1002/chem.201001123 |
| 50. | Arcadi, A.; Abbiati, G.; Rossi, E. J. Organomet. Chem. 2011, 696, 87–98. doi:10.1016/j.jorganchem.2010.08.011 |
| 51. | Kitahara, H.; Sakurai, H. J. Organomet. Chem. 2011, 696, 442–449. doi:10.1016/j.jorganchem.2010.08.038 |
| 19. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 20. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 21. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 22. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 23. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 24. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 25. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k |
| 26. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 27. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 28. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088 |
| 55. | Ochida, A.; Ito, H.; Sawamura, M. J. Am. Chem. Soc. 2006, 128, 16486–16487. doi:10.1021/ja066800c |
© 2011 Ito et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)