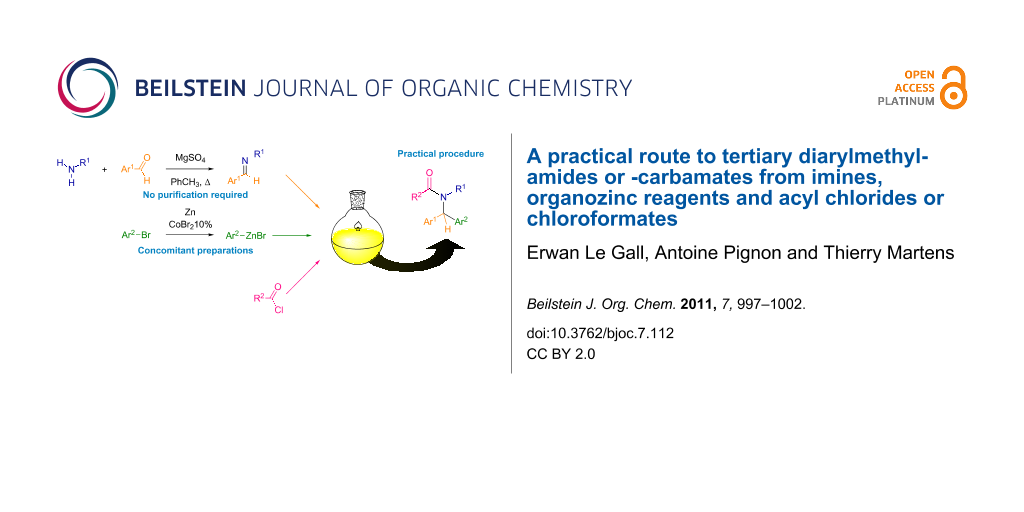
Abstract
A practical route to tertiary diarylmethylamides or -carbamates from imines, organozinc reagents and acyl chlorides or chloroformates is described. This route involves the formation of an imine, which is used without isolation, followed by its activation by the carbonyl-containing electrophile and the trapping of the acyliminium by an organozinc reagent. Most steps are conducted concomitantly to render the procedure as practical and straightforward as possible. Therefore, the whole experimental protocol takes less than two hours.

Graphical Abstract
Introduction
Diarylmethylamines constitute an important class of nitrogen-containing compounds displaying antihistaminic, anti-arrhythmic, diuretic, antidepressant, laxative, anesthetic and anticholinergic properties [1,2]. In this context, diarylmethylamides and -carbamates represent reliable N-protected diarylmethylamine derivatives and should thus serve as valuable precursors in the preparation of compounds of pharmaceutical interest. Several procedures enabling the construction of the diarylmethylamide and -carbamate core have been described. However, with respect to the substitution pattern of the expected final compound, available methods differ notably. Indeed, while the synthesis of secondary N-protected diarylmethylamines generally relies on the addition of organometallic reagents to electron-deficient (activated) imines [3-7], the preparation of tertiary diarylmethylamides or -carbamates may be conducted through the addition of aromatic nucleophiles onto N-acyliminium intermediates, formed in situ by reaction of imines with carbonyl-containing electrophiles. In this latter area, several studies have shown that some electron-enriched arenes can be used as nucleophiles and add efficiently onto the iminium carbon, either inter- or intramolecularly [8-16]. However, an increased range of aromatic moieties can be introduced through the use of organometallic compounds. The most commonly employed reagents are organoindium [17,18], organolithium [19,20], organomagnesium [21,22], organotin [23], or organozinc compounds [24,25]. However, although these are recognized as mild multi-purpose reagents, sole examples of their use in nucleophilic additions on acylimium salts consist, to the best of our knowledge, of the phenylation of quinolinium salts using diphenylzinc [26,27].
Recently, our group has been involved in various projects pertaining to the development of multicomponent reactions (MCRs) involving organometallic reagents, in particular organozinc reagents, due to their ability to react in very mild conditions and generally preserve most common functional groups. Moreover, used in stoichiometric amounts, organozinc reagents are more cost-effective and produce less toxic wastes than other common nucleophiles, such as, e.g., organoindium or organotin reagents. Our main contribution to the field was with regards to the use of arylzinc reagents in Mannich-type reactions with secondary amines and aldehydes to furnish tertiary diarylmethylamines [28-33]. However, while a large range of starting compounds could be used successfully in the process, we noticed that primary amines are ineffective, probably due to a weaker electrophilicity of the in situ-formed imines compared to iminium ions. Consequently, we report herein the use of primary amines in a sequential one-pot process, based on the preliminary activation of an aldimine with an acyl chloride or a chloroformate, and the subsequent trapping of the resulting acyliminium ion with an aromatic organozinc reagent, to generate a range of diarylmethylamides and -carbamates in satisfactory to good yields.
Results and Discussion
The limited intrinsic reactivity of imines towards the addition of nucleophiles has long been recognized as a major issue in nitrogen chemistry, but one which can be circumvented through several strategies, mainly intended to withdraw electrons and render the carbon more electrophilic [3-7]. Depending on the substitution pattern of the expected final amines, the increase of the electrophilicity should be implemented through the use of activated imines (Scheme 1, pathway A) or by quaternarization of the nitrogen atom with an electrophilic species (Scheme 1, pathway B). The activating group (AG) should then be released by a final deprotection to deliver the free amine (Scheme 1).
![[1860-5397-7-112-i1]](/bjoc/content/inline/1860-5397-7-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Addition of nucleophiles onto activated imines (A) or iminiums (B).
Scheme 1: Addition of nucleophiles onto activated imines (A) or iminiums (B).
During the course of preceding works, we noticed that the addition of aromatic organozinc reagents onto N-substituted aldimines, formed in situ upon reaction of primary amines with aromatic aldehydes, cannot be undertaken under our established conditions. Thus, we intended to activate the C=N double bond by rendering the carbon more electrophilic and we initially envisaged the use of Lewis acid catalysis. Indeed, we assumed that under these conditions, the formation of N–AG (AG = activating group) bonds would be reversible, thus cleavage would be effective in situ and only relatively small amounts of the Lewis acid would be necessary. While several common Lewis acids (TiCl4, AlCl3, CeCl3 and BF3·Et2O) were trialled unsuccessfully, a different strategy based on the formation of solid bonds indicated that carbonyl derivatives such as acetyl chloride or methyl chloroformate were, in contrast, efficient activators of the C=N double bond, albeit used in stoichiometric amounts. This result is consistent with some previous studies reporting the activation of imines under an acyliminium form and the subsequent addition of either aromatic [17-23,26,27] or non-aromatic [34,35] organometallic nucleophiles onto carbon.
Our preliminary investigations were then conducted on N-benzylidenepropan-1-amine, taken as a model aldimine, which was preformed and purified prior to use. This compound was subjected to consecutive reactions with acetyl chloride and phenylzinc bromide, furnishing the corresponding diarylmethylamide in good yield (80%). However, as supplementary experiments indicated that the starting imine 1 can be used without preliminary purification, we chose to simplify the process by operating from the crude imine, although slightly lower yields (10–15% decreasing) were hence obtained. Thus, in a typical experiment, the amine and the aldehyde were heated in toluene in the presence of 4 Å molecular sieves for a few minutes. After cooling to room temperature, the toluene solution containing the imine 1 was transferred into another flask in which a slight excess of the electrophile (acyl chloride or chloroformate 2) was added. After a limited period under heating, the arylzinc reagent 3, prepared in parallel via a cobalt-catalyzed procedure [36] was added and the resulting solution was stirred for 30 minutes at ambient temperature. The chromatographic purification of the crude oil afforded the expected diarylmethylamide or -carbamate 4. Representative experimental results are reported in Table 1.
Table 1: Formation of diarylmethylamides and -carbamates.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-112-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | R3 | R4 | Product | Isolated yield (%) |
|---|---|---|---|---|---|---|
| 1 | n-Pr | Ph | Me | Ph |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-112-i4.svg?max-width=637&scale=1.0)
4a |
64 |
| 2 | n-Pr | 3-O2N–C6H4– | Me | Ph |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-112-i5.svg?max-width=637&scale=1.0)
4b |
55 |
| 3 | n-Pr | 3-O2N–C6H4– | MeO | Ph |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-112-i6.svg?max-width=637&scale=1.0)
4c |
59 |
| 4 | n-Pr | 2-F3C–C6H4– | MeO | Ph |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-112-i7.svg?max-width=637&scale=1.0)
4d |
67 |
| 5 | n-Pr | thiophen-3-yl | MeO | Ph |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-112-i8.svg?max-width=637&scale=1.0)
4e |
42 |
| 6 | n-Pr | Ph | MeO | 3-F3C–C6H4– |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-112-i9.svg?max-width=637&scale=1.0)
4f |
68 |
| 7 | n-Pr | Ph | MeO | 4-EtO2C–C6H4– |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-112-i10.svg?max-width=637&scale=1.0)
4g |
57 |
| 8 | n-Pr | Ph | MeO | 4-Cl–C6H4– |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-112-i11.svg?max-width=637&scale=1.0)
4h |
71 |
| 9 | n-Pr | Ph | MeO | 4-MeO–C6H4– |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-112-i12.svg?max-width=637&scale=1.0)
4i |
63 |
| 10 | Ph | Ph | Me | Ph |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-112-i13.svg?max-width=637&scale=1.0)
4j |
69 |
| 11 | Bn | Ph | Me | Ph |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-112-i14.svg?max-width=637&scale=1.0)
4k |
36 |
aExperiments were conducted with ~10 mmol of imine, 12 mmol of acyl chloride or chloroformate, 13–16 mmol of the organozinc reagent, prepared from 20 mmol of aryl bromide.
Under these conditions, coupling products 4 are formed in low to high yields. The use of acetyl chloride (Table 1, entries 1, 2, 10 and 11) provided similar results to those observed with methyl chloroformate (Table 1, entries 3–9). It can be seen that more limited yields were obtained when a thiophene-derived aldehyde (Table 1, entry 5) or benzylamine was employed as the starting amine (Table 1, entry 11). However, these last two results could not be explained.
We next tried to extend the reaction to other electrophilic compounds that are known to easily form N–AG bonds with imines and furnish analogous iminium salts. The case of methanesulfonyl chloride (MsCl) was dealt with first (Scheme 2).
![[1860-5397-7-112-i2]](/bjoc/content/inline/1860-5397-7-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Activation of the aldimine with MsCl.
Scheme 2: Activation of the aldimine with MsCl.
Unfortunately, while the reaction of benzylzinc bromide proved efficient under MsCl activation, phenylzinc bromide did not undergo the coupling at all [37]. This was also the case with trimethylsilyl chloride as an activator, whose reaction with the model aldimine and phenylzinc bromide did not afford the expected compound. On the other hand, a preliminary experiment indicated that trifluoracetic anhydride was a very reliable activator of the imine towards phenylzinc bromide addition. These results, combined with the above reported observations with common Lewis acids, may indicate that acylating reagents are particularly reliable for the activation of aldimines toward arylzinc additions. However, the use of carbonyl-containing electrophiles obviously constitutes an important drawback of the procedure. Indeed, although TBAF has been reported to constitute a mild deprotection reagent for a range of carbamates [38,39], the cleavage of the N–C=O bond, which is formed during the process, might be commonly achieved under rather harsh conditions. This is not the case with other potential activating agents such as sulfinic or phosphinic chloride derivatives (ClS(O)R and ClP(O)R2), whose N–AG bond might be cleaved easily upon acidic work-up. In addition, chiral versions of such activators would be of further interest for potential asymmetric couplings (at least with benzylzinc reagents), as the stereogenic center would be located very close to the impending asymmetric carbon and may thus serve as a valuable chiral auxiliary. Consequently, we envisage the implementation a further study which would be dedicated to the evaluation of well-recognized chiral inductors such as Ellman- [40,41] or Davis-type [42,43] sulfinyl derivatives in the process.
Conclusion
In conclusion, the results reported in this study indicate that the formation of acyliminium cations constitutes a very convenient approach to the activation of imines toward the addition of aromatic organozinc reagents. Indeed, we could prepare a range of diarylmethylamides or diarylmethylcarbamates by a sequential multicomponent process involving the preliminary formation of an imine, which can be used without isolation, its activation by an acyl chloride or a chloroformate and the final trapping of the resulting acyliminium salt by an arylzinc reagent. However, the harsh conditions which would probably be required for the deprotection of the amide or carbamate function prompt us to undertake complementary experiments dedicated to the assessment of easier-to-cleave activating groups. Consequently, the evaluation of sulfinyl- or phosphinyl derivatives in the process has been undertaken recently and will be reported in due course.
Experimental
Typical procedure for the preparation of diarylmethylamides and carbamates
The aldimine (~10 mmol) was prepared from the aromatic aldehyde (12 mmol) and the amine (12 mmol) in toluene (10 mL) in the presence of 4 Å molecular sieves (10 g) and para-toluenesulfonic acid (10 mg). After 30 min stirring at 80 °C and cooling to rt, the solution was taken-up with a syringe and the sieves washed with 5 mL toluene. The combined toluene fractions were placed in another flask, which was flushed with argon prior to addition, and acetyl chloride or methyl chloroformate (12 mmol) was added. The resulting mixture was stirred at rt (ClCOCH3) or at 50 °C (ClCOOCH3) for 30 min, a period during which the aromatic organozinc reagent (13–16 mmol, depending on the starting halide) was prepared concomitantly as follows: A 100 mL round bottom flask was flushed with argon, then acetonitrile (20 mL), zinc dust (3 g), TFA (0.2 mL) and BrCH2CH2Br (0.2 mL) were added consecutively under vigorous (~500 rpm) stirring. The mixture was heated until gas was evolved (at 50–70 °C), then allowed to cool to rt under continuous stirring. The aryl bromide (15 mmol) and anhydrous cobalt bromide (330 mg) were then added to the mixture, which was stirred at rt for additional 20 min. Stirring was then stopped and the surrounding solution was taken-up with a syringe. The solution was then added to the flask containing the imine/carbonyl-containing compound mixture and the resulting mixture was stirred at rt for 30 min. The solution was poured into a sat. NH4Cl solution (100 mL), extracted with diethyl ether (2 × 75 mL) and the combined organic fractions were dried with magnesium sulfate, filtrated and then concentrated under reduced pressure. The crude oil was purified by column chromatography over silica gel with a pentane/diethyl ether mixture (1:0 to 0:1) as an eluant to afford the diarylmethylamide or carbamate 4.
NMR data for selected compounds
Methyl phenyl(2-(trifluoromethyl)phenyl)methyl(propyl)carbamate (4d) 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.8 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.37–7.25 (m, 4H), 7.12 (d, J = 7.2 Hz, 2H), 6.94 (s, 1H), 3.71 (s, 3H), 3.45–3.31 (m, 1H), 3.23–3.11 (m, 1H), 1.27–1.11 (m, 1H), 0.79 (br s, 1H), 0.56 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.9, 139.9, 139.5, 131.7, 131.0, 129.5 (q, J = 30.3 Hz), 128.4, 128.0, 127.9, 127.4, 126.4 (q, J = 6.0 Hz), 124.2 (q, J = 274.4 Hz), 59.4, 52.7, 47.5, 21.9, 11.11.
N-Benzhydryl-N-phenylacetamide (4j) 1H NMR (400 MHz, CDCl3) δ 7.19–7.10 (m, 15H), 6.74 (s, 1H), 1.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.8, 140.9, 139.2, 130.2, 129.7, 128.9, 128.1, 128.0, 127.4, 64.1, 23.7.
N-Benzhydryl-N-benzylacetamide (4k) 1H NMR (400 MHz, CDCl3) δ 7.15–6.96 (m, 14H), 6.67–6.64 (m, 2H), 4.57 (s, 2H), 2.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.2, 139.3, 137.4, 129.2, 128.5, 128.2, 127.9, 127.6, 125.8, 66.4, 48.0, 22.8.
References
-
Spencer, C. M.; Faulds, D.; Peters, D. H. Drugs 1993, 46, 1055–1080.
Return to citation in text: [1] -
Sakurai, S.; Ogawa, N.; Suzuki, T.; Kato, K.; Ohashi, T.; Yasuda, S.; Kato, H.; Ito, Y. Chem. Pharm. Bull. 1996, 44, 765–777.
Return to citation in text: [1] -
Enders, D.; Reinhold, U. Tetrahedron: Asymmetry 1997, 8, 1895–1946. doi:10.1016/S0957-4166(97)00208-5
Return to citation in text: [1] [2] -
Bloch, R. Chem. Rev. 1998, 98, 1407–1438. doi:10.1021/cr940474e
Return to citation in text: [1] [2] -
Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z
Return to citation in text: [1] [2] -
Friestad, G. K.; Mathies, A. K. Tetrahedron 2007, 63, 2541–2569. doi:10.1016/j.tet.2006.11.076
Return to citation in text: [1] [2] -
Hermanns, N.; Dahmen, S.; Bolm, C.; Bräse, S. Angew. Chem. 2002, 114, 3844–3846. doi:10.1002/1521-3757(20021004)114:19<3844::AID-ANGE3844>3.0.CO;2-R
Return to citation in text: [1] [2] -
Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387
Return to citation in text: [1] -
Kitabatake, M.; Saitoh, T.; Sano, T.; Horiguchi, Y. Heterocycles 2009, 78, 1177–1181. doi:10.3987/COM-08-11621
Return to citation in text: [1] -
Zhang, Y.; DeSchepper, D. J.; Gilbert, T. M.; Sai, K. K. S.; Klumpp, D. A. Chem. Commun. 2007, 4032–4034. doi:10.1039/b708760h
Return to citation in text: [1] -
Romero, M.; Caignard, D.-H.; Renard, P.; Pujol, M. D. Tetrahedron 2008, 64, 11020–11027. doi:10.1016/j.tet.2008.09.097
Return to citation in text: [1] -
Klumpp, D. A.; Zhang, Y.; O’Connor, M. J.; Esteves, P. M.; de Almeida, L. S. Org. Lett. 2007, 9, 3085–3088. doi:10.1021/ol0711570
Return to citation in text: [1] -
Venkov, A. P.; Statkova-Abeghe, S. Synth. Commun. 1996, 26, 127–134. doi:10.1080/00397919608003871
Return to citation in text: [1] -
Venkov, A. P.; Statkova, S. M.; Ivanov, I. I. Synth. Commun. 1992, 22, 125–134. doi:10.1080/00397919208021083
Return to citation in text: [1] -
Venkov, A. P.; Statkova, S. M. Synth. Commun. 1991, 21, 1511–1520. doi:10.1080/00397919108016425
Return to citation in text: [1] -
Venkov, A. P.; Mollov, N. M. Synthesis 1982, 216–217. doi:10.1055/s-1982-29752
Return to citation in text: [1] -
Black, D. A.; Arndtsen, B. A. Org. Lett. 2006, 8, 1991–1993. doi:10.1021/ol060277p
Return to citation in text: [1] [2] -
Beveridge, R. E.; Black, D. A.; Arndtsen, B. A. Eur. J. Org. Chem. 2010, 3650–3656. doi:10.1002/ejoc.201000231
Return to citation in text: [1] [2] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014
Return to citation in text: [1] [2] -
Alexakis, A.; Amiot, F. Tetrahedron: Asymmetry 2002, 13, 2117–2122. doi:10.1016/S0957-4166(02)00531-1
Return to citation in text: [1] [2] -
Venkov, A. P.; Statkova-Abeghe, S. M. Synth. Commun. 1995, 1817–1824. doi:10.1080/00397919508015425
Return to citation in text: [1] [2] -
Pegoraro, S.; Lang, M.; Dreker, T.; Kraus, J.; Hamm, S.; Meere, C.; Feurle, J.; Tasler, S.; Prütting, S.; Kuras, Z.; Visan, V.; Grissmer, S. Bioorg. Med. Chem. Lett. 2009, 19, 2299–2304. doi:10.1016/j.bmcl.2009.02.077
Return to citation in text: [1] [2] -
Black, D. A.; Arndtsen, B. A. J. Org. Chem. 2005, 70, 5133–5138. doi:10.1021/jo0503557
Return to citation in text: [1] [2] -
Knochel, P.; Singer, R. D. Chem. Rev. 1993, 93, 2117–2188. doi:10.1021/cr00022a008
Return to citation in text: [1] -
Knochel, P.; Jones, P., Eds. Organozinc Reagents, A Practical Approach; The Practical Approach in Chemistry Series; Oxford University Press: Oxford, 1999.
Return to citation in text: [1] -
Ludwig, M.; Polborn, K.; Wanner, K. T. Heterocycles 2003, 61, 299–326. doi:10.3987/COM-03-S40
Return to citation in text: [1] [2] -
Bender, C.; Liebscher, J. ARKIVOC 2009, (vi), 111–136.
Return to citation in text: [1] [2] -
Haurena, C.; Le Gall, E.; Sengmany, S.; Martens, T. Tetrahedron 2010, 66, 9902–9911. doi:10.1016/j.tet.2010.10.058
Return to citation in text: [1] -
Le Gall, E.; Haurena, C.; Sengmany, S.; Martens, T.; Troupel, M. J. Org. Chem. 2009, 74, 7970–7973. doi:10.1021/jo901704s
Return to citation in text: [1] -
Sengmany, S.; Le Gall, E.; Troupel, M. Synlett 2008, 1031–1035.
Return to citation in text: [1] -
Sengmany, S.; Le Gall, E.; Le Jean, C.; Troupel, M.; Nédélec, J.-Y. Tetrahedron 2007, 63, 3672–3681. doi:10.1016/j.tet.2007.02.086
Return to citation in text: [1] -
Le Gall, E.; Troupel, M.; Nédélec, J.-Y. Tetrahedron 2006, 62, 9953–9965. doi:10.1016/j.tet.2006.08.008
Return to citation in text: [1] -
Le Gall, E.; Troupel, M.; Nédélec, J.-Y. Tetrahedron Lett. 2006, 47, 2497–2500. doi:10.1016/j.tetlet.2006.02.056
Return to citation in text: [1] -
Black, D. A.; Arndtsen, B. A. Org. Lett. 2004, 6, 1107–1110. doi:10.1021/ol036462+
Return to citation in text: [1] -
Fischer, C.; Carreira, E. M. Org. Lett. 2004, 6, 1497–1499. doi:10.1021/ol049578u
Return to citation in text: [1] -
Fillon, H.; Gosmini, C.; Périchon, J. J. Am. Chem. Soc. 2003, 125, 3867–3870. doi:10.1021/ja0289494
Return to citation in text: [1] -
The more important reactivity of benzylzinc vs arylzinc reagents has been noticed on several occasions. For instance, we have already shown that benzyl bromides react, under Barbier-like conditions, with aldehydes and primary amines whereas aryl bromides do not undergo the coupling at all. This result is consistent with a possible nucleophilic addition of benzylzinc reagents onto imines without mandatory activation of the latter. See ref. [29] for details.
Return to citation in text: [1] -
Routier, S.; Saugé, L.; Ayerbe, N.; Coudert, G.; Mérour, J.-Y. Tetrahedron Lett. 2002, 43, 589–591. doi:10.1016/S0040-4039(01)02225-0
Return to citation in text: [1] -
Jacquemard, U.; Bénéteau, V.; Lefoix, M.; Routier, S.; Mérour, J.-Y.; Coudert, G. Tetrahedron 2004, 60, 10039–10047. doi:10.1016/j.tet.2004.07.071
Return to citation in text: [1] -
Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z
Return to citation in text: [1] -
Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u
Return to citation in text: [1] -
Davis, F. A. J. Org. Chem. 2006, 71, 8993–9003. doi:10.1021/jo061027p
Return to citation in text: [1] -
Zhou, P.; Chen, B.-C.; Davis, F. A. Tetrahedron 2004, 60, 8003–8030. doi:10.1016/j.tet.2004.06.071
Return to citation in text: [1]
| 29. | Le Gall, E.; Haurena, C.; Sengmany, S.; Martens, T.; Troupel, M. J. Org. Chem. 2009, 74, 7970–7973. doi:10.1021/jo901704s |
| 40. | Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913–9914. doi:10.1021/ja972012z |
| 41. | Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u |
| 42. | Davis, F. A. J. Org. Chem. 2006, 71, 8993–9003. doi:10.1021/jo061027p |
| 43. | Zhou, P.; Chen, B.-C.; Davis, F. A. Tetrahedron 2004, 60, 8003–8030. doi:10.1016/j.tet.2004.06.071 |
| 1. | Spencer, C. M.; Faulds, D.; Peters, D. H. Drugs 1993, 46, 1055–1080. |
| 2. | Sakurai, S.; Ogawa, N.; Suzuki, T.; Kato, K.; Ohashi, T.; Yasuda, S.; Kato, H.; Ito, Y. Chem. Pharm. Bull. 1996, 44, 765–777. |
| 19. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014 |
| 20. | Alexakis, A.; Amiot, F. Tetrahedron: Asymmetry 2002, 13, 2117–2122. doi:10.1016/S0957-4166(02)00531-1 |
| 37. | The more important reactivity of benzylzinc vs arylzinc reagents has been noticed on several occasions. For instance, we have already shown that benzyl bromides react, under Barbier-like conditions, with aldehydes and primary amines whereas aryl bromides do not undergo the coupling at all. This result is consistent with a possible nucleophilic addition of benzylzinc reagents onto imines without mandatory activation of the latter. See ref. [29] for details. |
| 17. | Black, D. A.; Arndtsen, B. A. Org. Lett. 2006, 8, 1991–1993. doi:10.1021/ol060277p |
| 18. | Beveridge, R. E.; Black, D. A.; Arndtsen, B. A. Eur. J. Org. Chem. 2010, 3650–3656. doi:10.1002/ejoc.201000231 |
| 38. | Routier, S.; Saugé, L.; Ayerbe, N.; Coudert, G.; Mérour, J.-Y. Tetrahedron Lett. 2002, 43, 589–591. doi:10.1016/S0040-4039(01)02225-0 |
| 39. | Jacquemard, U.; Bénéteau, V.; Lefoix, M.; Routier, S.; Mérour, J.-Y.; Coudert, G. Tetrahedron 2004, 60, 10039–10047. doi:10.1016/j.tet.2004.07.071 |
| 8. | Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387 |
| 9. | Kitabatake, M.; Saitoh, T.; Sano, T.; Horiguchi, Y. Heterocycles 2009, 78, 1177–1181. doi:10.3987/COM-08-11621 |
| 10. | Zhang, Y.; DeSchepper, D. J.; Gilbert, T. M.; Sai, K. K. S.; Klumpp, D. A. Chem. Commun. 2007, 4032–4034. doi:10.1039/b708760h |
| 11. | Romero, M.; Caignard, D.-H.; Renard, P.; Pujol, M. D. Tetrahedron 2008, 64, 11020–11027. doi:10.1016/j.tet.2008.09.097 |
| 12. | Klumpp, D. A.; Zhang, Y.; O’Connor, M. J.; Esteves, P. M.; de Almeida, L. S. Org. Lett. 2007, 9, 3085–3088. doi:10.1021/ol0711570 |
| 13. | Venkov, A. P.; Statkova-Abeghe, S. Synth. Commun. 1996, 26, 127–134. doi:10.1080/00397919608003871 |
| 14. | Venkov, A. P.; Statkova, S. M.; Ivanov, I. I. Synth. Commun. 1992, 22, 125–134. doi:10.1080/00397919208021083 |
| 15. | Venkov, A. P.; Statkova, S. M. Synth. Commun. 1991, 21, 1511–1520. doi:10.1080/00397919108016425 |
| 16. | Venkov, A. P.; Mollov, N. M. Synthesis 1982, 216–217. doi:10.1055/s-1982-29752 |
| 34. | Black, D. A.; Arndtsen, B. A. Org. Lett. 2004, 6, 1107–1110. doi:10.1021/ol036462+ |
| 35. | Fischer, C.; Carreira, E. M. Org. Lett. 2004, 6, 1497–1499. doi:10.1021/ol049578u |
| 3. | Enders, D.; Reinhold, U. Tetrahedron: Asymmetry 1997, 8, 1895–1946. doi:10.1016/S0957-4166(97)00208-5 |
| 4. | Bloch, R. Chem. Rev. 1998, 98, 1407–1438. doi:10.1021/cr940474e |
| 5. | Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z |
| 6. | Friestad, G. K.; Mathies, A. K. Tetrahedron 2007, 63, 2541–2569. doi:10.1016/j.tet.2006.11.076 |
| 7. | Hermanns, N.; Dahmen, S.; Bolm, C.; Bräse, S. Angew. Chem. 2002, 114, 3844–3846. doi:10.1002/1521-3757(20021004)114:19<3844::AID-ANGE3844>3.0.CO;2-R |
| 36. | Fillon, H.; Gosmini, C.; Périchon, J. J. Am. Chem. Soc. 2003, 125, 3867–3870. doi:10.1021/ja0289494 |
| 26. | Ludwig, M.; Polborn, K.; Wanner, K. T. Heterocycles 2003, 61, 299–326. doi:10.3987/COM-03-S40 |
| 27. | Bender, C.; Liebscher, J. ARKIVOC 2009, (vi), 111–136. |
| 3. | Enders, D.; Reinhold, U. Tetrahedron: Asymmetry 1997, 8, 1895–1946. doi:10.1016/S0957-4166(97)00208-5 |
| 4. | Bloch, R. Chem. Rev. 1998, 98, 1407–1438. doi:10.1021/cr940474e |
| 5. | Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z |
| 6. | Friestad, G. K.; Mathies, A. K. Tetrahedron 2007, 63, 2541–2569. doi:10.1016/j.tet.2006.11.076 |
| 7. | Hermanns, N.; Dahmen, S.; Bolm, C.; Bräse, S. Angew. Chem. 2002, 114, 3844–3846. doi:10.1002/1521-3757(20021004)114:19<3844::AID-ANGE3844>3.0.CO;2-R |
| 24. | Knochel, P.; Singer, R. D. Chem. Rev. 1993, 93, 2117–2188. doi:10.1021/cr00022a008 |
| 25. | Knochel, P.; Jones, P., Eds. Organozinc Reagents, A Practical Approach; The Practical Approach in Chemistry Series; Oxford University Press: Oxford, 1999. |
| 17. | Black, D. A.; Arndtsen, B. A. Org. Lett. 2006, 8, 1991–1993. doi:10.1021/ol060277p |
| 18. | Beveridge, R. E.; Black, D. A.; Arndtsen, B. A. Eur. J. Org. Chem. 2010, 3650–3656. doi:10.1002/ejoc.201000231 |
| 19. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014 |
| 20. | Alexakis, A.; Amiot, F. Tetrahedron: Asymmetry 2002, 13, 2117–2122. doi:10.1016/S0957-4166(02)00531-1 |
| 21. | Venkov, A. P.; Statkova-Abeghe, S. M. Synth. Commun. 1995, 1817–1824. doi:10.1080/00397919508015425 |
| 22. | Pegoraro, S.; Lang, M.; Dreker, T.; Kraus, J.; Hamm, S.; Meere, C.; Feurle, J.; Tasler, S.; Prütting, S.; Kuras, Z.; Visan, V.; Grissmer, S. Bioorg. Med. Chem. Lett. 2009, 19, 2299–2304. doi:10.1016/j.bmcl.2009.02.077 |
| 23. | Black, D. A.; Arndtsen, B. A. J. Org. Chem. 2005, 70, 5133–5138. doi:10.1021/jo0503557 |
| 26. | Ludwig, M.; Polborn, K.; Wanner, K. T. Heterocycles 2003, 61, 299–326. doi:10.3987/COM-03-S40 |
| 27. | Bender, C.; Liebscher, J. ARKIVOC 2009, (vi), 111–136. |
| 23. | Black, D. A.; Arndtsen, B. A. J. Org. Chem. 2005, 70, 5133–5138. doi:10.1021/jo0503557 |
| 21. | Venkov, A. P.; Statkova-Abeghe, S. M. Synth. Commun. 1995, 1817–1824. doi:10.1080/00397919508015425 |
| 22. | Pegoraro, S.; Lang, M.; Dreker, T.; Kraus, J.; Hamm, S.; Meere, C.; Feurle, J.; Tasler, S.; Prütting, S.; Kuras, Z.; Visan, V.; Grissmer, S. Bioorg. Med. Chem. Lett. 2009, 19, 2299–2304. doi:10.1016/j.bmcl.2009.02.077 |
| 28. | Haurena, C.; Le Gall, E.; Sengmany, S.; Martens, T. Tetrahedron 2010, 66, 9902–9911. doi:10.1016/j.tet.2010.10.058 |
| 29. | Le Gall, E.; Haurena, C.; Sengmany, S.; Martens, T.; Troupel, M. J. Org. Chem. 2009, 74, 7970–7973. doi:10.1021/jo901704s |
| 30. | Sengmany, S.; Le Gall, E.; Troupel, M. Synlett 2008, 1031–1035. |
| 31. | Sengmany, S.; Le Gall, E.; Le Jean, C.; Troupel, M.; Nédélec, J.-Y. Tetrahedron 2007, 63, 3672–3681. doi:10.1016/j.tet.2007.02.086 |
| 32. | Le Gall, E.; Troupel, M.; Nédélec, J.-Y. Tetrahedron 2006, 62, 9953–9965. doi:10.1016/j.tet.2006.08.008 |
| 33. | Le Gall, E.; Troupel, M.; Nédélec, J.-Y. Tetrahedron Lett. 2006, 47, 2497–2500. doi:10.1016/j.tetlet.2006.02.056 |
© 2011 Le Gall et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)