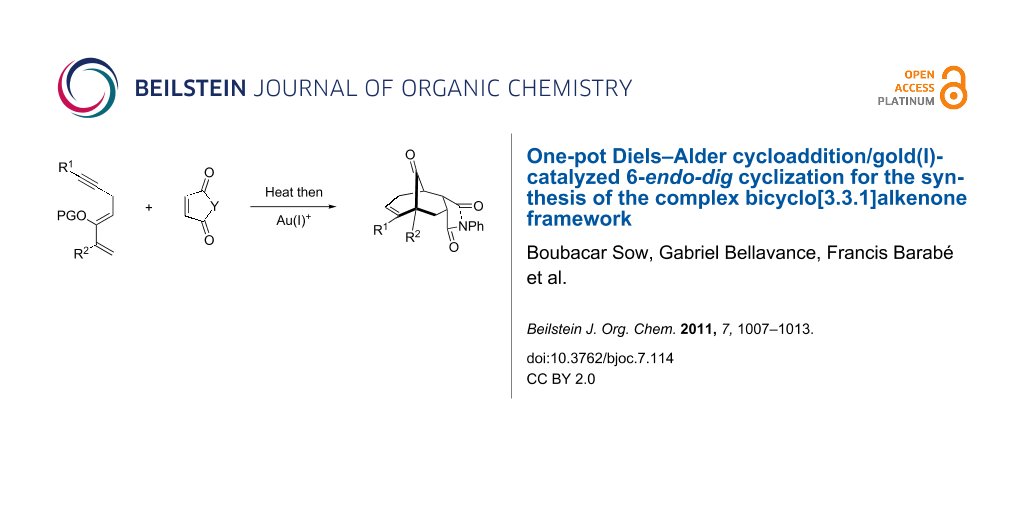
Abstract
The rapid synthesis of bicyclo[m.n.1]alkanone cores possessing quaternary carbon centers adjacent to a bridged ketone represents a significant synthetic challenge. This type of architectural feature is embedded in various complex biologically active compounds such as hyperforin and garsubellin A. Herein, we report a highly diastereoselective one-pot Diels–Alder reaction/Au(I)-catalyzed carbocyclization to generate bicyclo[3.3.1]alkanones in yields ranging from 48–93%.

Graphical Abstract
Introduction
Highly oxygenated and densely substituted carbon-bridged medium sized rings such as 1 are commonly found in nature as structural frameworks of many important bioactive natural products, and in particular, polycyclic polyprenylated acetylphloroglucinols (PPAPs) (Figure 1) [1]. In the past decades, more than 100 PPAPs exhibiting a wide variety of biological activities (antibiotic, anti-HIV, anti-oxidant, etc.) have been isolated from Guttiferrea plants such as hyperforin (2) [2-6] and garsubellin A (3) [7,8]. The challenging synthesis of PPAP structures combined with their promising therapeutic potential has drawn attention from several research groups [9-12].
![[1860-5397-7-114-1]](/bjoc/content/figures/1860-5397-7-114-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of naturally occurring PPAPs.
Figure 1: Structures of naturally occurring PPAPs.
In 2009, we reported a mild and highly efficient method to generate carbon-bridged frameworks of various sizes through a gold(I)-catalyzed carbocyclization [13]. Although the cyclization of enol ether 5 can produce 5-exo and 6-endo products, we found that gold complexes 6, having bulky phosphine ligands such as 2-bis(tert-butylphosphino)biphenyl, gave exclusively the 6-endo-dig cyclized products 7 (Scheme 1). In the course of our studies directed towards the synthesis of naturally occurring PPAPs and related carbon-bridged ketone scaffolds, we envisioned that PPAP framework 1 could be generated via a Au(I)-catalyzed cyclization [14-22].
![[1860-5397-7-114-i1]](/bjoc/content/inline/1860-5397-7-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Gold(I)-catalyzed 6-endo-dig cyclization.
Scheme 1: Gold(I)-catalyzed 6-endo-dig cyclization.
Results and Discussion
The synthesis began by a C-alkylation of enone 8 [23] using LDA and MeI to give the corresponding ketone in 90% yield (Scheme 2). A second alkylation to add the propargyl chain was carried out using LDA and propargyl bromide to afford 9 in 62% yield as an inseparable mixture of diastereomers (dr = 3:1). Subsequently, conjugate addition of methylmagnesium bromide in the presence of a catalytic amount of CuI provided the corresponding ketone in 53% yield. The ketone was then treated with TBSCl, NaI and triethylamine to give the desired silylenol ether 10 in 46% yield, which upon exposure to the Au(I) complex 6 (2 mol %) provided the desired bicyclo[3.3.1]nonenone 11 in 88% yield. It is important to note that the Au(I)-catalyzed cyclization proceeds in high yields in a sterically congested environment. The synthesis of the core of papuaforin (11) was achieved in five steps from enone 8.
![[1860-5397-7-114-i2]](/bjoc/content/inline/1860-5397-7-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of papuaforin A core 4.
Scheme 2: Synthesis of papuaforin A core 4.
However, one might recognize that the low chemical yields encountered in some steps undermine the efficacy of the Au(I)-catalyzed cyclization approach. In order to solve this issue, we assumed that bicyclo[3.3.1]nonenone scaffolds can be directly obtained through an intermolecular Diels–Alder reaction/Au(I)-catalyzed 6-endo-dig carbocyclization (Scheme 3). Cycloaddition between diene 12 and dienophile 13 should provide the endo cycloadduct 14, which, in the presence of a gold(I) catalyst, would form the gold complex A. This undergoes a carbocyclization of enol ether [24-31] to afford intermediate B, which after proto-deauration and hydrolysis affords the bridgehead ketone 15. The attractive feature of this process resides in the ability to generate four new stereogenic centers and three new C–C bonds in one single operation.
![[1860-5397-7-114-i3]](/bjoc/content/inline/1860-5397-7-114-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed domino Diels–Alder reaction/gold(I)-catalyzed cyclization.
Scheme 3: Proposed domino Diels–Alder reaction/gold(I)-catalyzed cyclization.
To validate the above hypothesis, diene 16 (Z-isomer) was heated with N-phenylmaleimide in toluene at 150 °C, for two hours, by microwave irradiation (Scheme 4) (see Supporting Information File 1 for experimental procedures). The solution containing the Diels–Alder adduct 17 was cooled down to room temperature and 2 mol % of Au(I) complex 6 was added. The bridgedhead ketone 18 was obtained in 80% yield as a single diastereomer. The relative stereochemistry of 18 was unambiguously established by X-ray analysis (see Supporting Information File 2). With this result in hand, we explored the scope of this sequential reaction (Table 1).
![[1860-5397-7-114-i4]](/bjoc/content/inline/1860-5397-7-114-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: One-pot Diels–Alder cycloaddition/gold(I) catalyzed carbocyclization.
Scheme 4: One-pot Diels–Alder cycloaddition/gold(I) catalyzed carbocyclization.
Table 1: Results of the one-pot Diels–Alder reaction/Au(I)-catalyzed cyclization.
| entry | diene | dienophile | product (yield)a |
|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-114-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-114-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-114-i7.svg?max-width=637&scale=1.0)
(93%) |
| 2 | 19 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-114-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-114-i9.svg?max-width=637&scale=1.0)
(51%) |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-114-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-114-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-114-i12.svg?max-width=637&scale=1.0)
(88%) |
| 4 | 20 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-114-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-114-i14.svg?max-width=637&scale=1.0)
(50%) |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-114-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-114-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-114-i17.svg?max-width=637&scale=1.0)
(81%) |
| 6 | 21 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-114-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-114-i19.svg?max-width=637&scale=1.0)
(78%) |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-114-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-114-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-114-i22.svg?max-width=637&scale=1.0)
(77%) |
| 8 | 22 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-114-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-114-i24.svg?max-width=637&scale=1.0)
(48%) |
| 9 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-114-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-114-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-114-i27.svg?max-width=637&scale=1.0)
(56%) |
aIsolated yield and dr > 25:1 in all cases.
One-pot cycloaddition/cyclization of dienes 19 and 20 (Z/E = 6:1 ca.) with N-phenylmaleimide gave ketones 24 and 26 in 93 and 88% yield, respectively, as the sole diastereomers (Table 1, entries 1 and 3). The use of maleic anhydride as the dienophile also provided the desired products 25 and 27, albeit in lower yields of 51 and 50%, respectively (Table 1, entries 2 and 4). Prenylated diene 21 was smoothly converted to ketones 28 and 29 in 81 and 78% yield, respectively (Table 1, entries 5 and 6). Table 1, entries 7 and 8 reveal that the diene 22, bearing a phenyl group at C2, can be stereoselectively transformed into the desired bridgehead ketones 30 and 31 in 77 and 48% yields, respectively. Interestingly, hemiketal 32 was isolated in 56% yield, which suggests that the MOM group was cleaved during the Au(I)-catalyzed carbocyclization. It is important to note that the E-isomer of dienes 19–23 (minor compound) do not react with the dienophiles, but rather isomerized to the Z-form under the reaction conditions, thus, ensuring the formation of a single diastereomer.
To extend the scope of the reaction, other dienes possessing internal alkynes were also investigated (Table 2). It can be seen that large substituents at the alkyne terminal position did not affect the efficacy of the reaction. Intermolecular cycloaddition/Au(I)-catalyzed cyclization of aryl acetylene dienes 33–35 provided the desired ketones 37–39 in yields ranging from 68 to 91% (Table 2, entries 1–3). Remarkably, enyne 36 was converted to 40 in 79% yield (Table 2, entry 4).
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-114-i28.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-114-i29.svg?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-114-i30.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-7-114-i31.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-7-114-i32.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-7-114-i33.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-7-114-i34.svg?max-width=637&scale=1.0)
![[Graphic 31]](/bjoc/content/inline/1860-5397-7-114-i35.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-7-114-i36.svg?max-width=637&scale=1.0)
Conclusion
In summary, we have developed an efficient stereoselective method for the construction of bicyclic[3.3.1]nonenone frameworks. This one-pot Diels–Alder/Au(I)-catalyzed carbocyclization process provides access to synthetically useful motifs that are found in numerous naturally occurring PPAPs. In addition, the Au(I)-catalyzed cyclization proved to be tolerant of a sterically crowded environment. Further studies to develop an enantioselective version of this reaction and its application to the total synthesis of hyperforin (2) and garsubellin A (3) are underway and will be reported in due course.
Acknowledgements
We thank the Natural Science and Engineering Research Council of Canada (NSERC), Merck Research Laboratories, Merck Frosst Canada, Boehringer Ingelheim (Laval), PREA, Canada Foundation for Innovation, Ontario Innovation Trust and the University of Ottawa for generous funding. F.B. thanks NSERC for post-graduate scholarship (CGS-D1).
References
-
Ciochina, R.; Grossman, R. B. Chem. Rev. 2006, 106, 3963. doi:10.1021/cr0500582
Return to citation in text: [1] -
Bystrov, N. S.; Chernov, B. K.; Dobrynin, V. N.; Kolosov, M. N. Tetrahedron Lett. 1975, 16, 2791. doi:10.1016/S0040-4039(00)75241-5
Return to citation in text: [1] -
Müller, W. E.; Singer, A.; Wonnemann, M.; Hafner, U.; Rolli, M.; Schäfer, C. Pharmacopsychiatry 1998, 31, 16. doi:10.1055/s-2007-979341
Return to citation in text: [1] -
Verotta, L.; Appendino, G.; Bombardelli, E.; Brun, R. Bioorg. Med. Chem. Lett. 2007, 17, 1544. doi:10.1016/j.bmcl.2006.12.100
Return to citation in text: [1] -
Gey, C.; Kyrylenko, S.; Henning, L.; Nguyen, L.-H. D.; Büttner, A.; Pham, H. D.; Giannis, A. Angew. Chem., Int. Ed. 2007, 46, 5219. doi:10.1002/anie.200605207
Return to citation in text: [1] -
Moore, L. B.; Goodwin, B.; Jones, S. A.; Wisely, G. B.; Serabjit-Singh, C. J.; Willson, T. M.; Collins, J. L.; Kliewer, S. A. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7500. doi:10.1073/pnas.130155097
Return to citation in text: [1] -
Fukuyama, Y.; Kuwayama, A.; Minami, H. Chem. Pharm. Bull. 1997, 45, 947.
Return to citation in text: [1] -
Fukuyama, Y.; Minami, H.; Kuwayama, A. Phytochemistry 1998, 49, 853. doi:10.1016/S0031-9422(98)00126-5
Return to citation in text: [1] -
Shimizu, Y.; Shi, S. L.; Usuda, H.; Kanai, M.; Shibasaki, M. Angew. Chem., Int. Ed. 2010, 49, 1103. doi:10.1002/anie.200906678
Return to citation in text: [1] -
Kuramochi, A.; Usuda, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 14200. doi:10.1021/ja055301t
Return to citation in text: [1] -
Danishefsky, S. J.; Siegel, D. R. J. Am. Chem. Soc. 2006, 128, 1048. doi:10.1021/ja057418n
Return to citation in text: [1] -
Ahmad, N. M.; Rodeschini, V.; Simpkins, N. S.; Ward, S. E.; Blake, A. J. J. Org. Chem. 2007, 72, 4803. doi:10.1021/jo070388h
Return to citation in text: [1] -
Barriault, L.; Barabé, F.; Bétournay, G.; Bellavance, G. Org. Lett. 2009, 11, 4236. doi:10.1021/ol901722q
Return to citation in text: [1] -
Toste, F. D.; Gorin, D. J. Nature 2007, 446, 395. doi:10.1038/nature05592
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333. doi:10.1039/B612008C
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d
Return to citation in text: [1] -
Skouta, R.; Li, C.-J. Tetrahedron 2008, 64, 4917. doi:10.1016/j.tet.2008.03.083
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 5, 675. doi:10.1055/s-0029-1219369
Return to citation in text: [1] -
Johnson, W. S.; McCarry, B. E.; Markezich, R. L.; Boots, S. G. J. Am. Chem. Soc. 1980, 102, 352. doi:10.1021/ja00521a057
Return to citation in text: [1] -
Dankwardt, J. W. Tetrahedron Lett. 2001, 42, 5809. doi:10.1016/S0040-4039(01)01146-7
Return to citation in text: [1] -
Suhre, M. H.; Reif, M.; Kirsch, S. F. Org. Lett. 2005, 7, 3925. doi:10.1021/ol0514101
Return to citation in text: [1] -
Staben, S. T.; Kennedy-Smith, J. J.; Huang, D.; Corkey, B. K.; LaLonde, R. L.; Toste, F. D. Angew. Chem., Int. Ed. 2006, 45, 5991. doi:10.1002/anie.200602035
Return to citation in text: [1] -
Linghu, X.; Kennedy-Smith, J. J.; Toste, F. D. Angew. Chem., Int. Ed. 2007, 46, 7671. doi:10.1002/anie.200702695
Return to citation in text: [1] -
Lee, K.; Lee, P. H. Adv. Synth. Catal. 2007, 349, 2092. doi:10.1002/adsc.200700304
Return to citation in text: [1] -
Minnihan, E. C.; Colletti, S. L.; Toste, F. D.; Shen, H. C. J. Org. Chem. 2007, 72, 6287. doi:10.1021/jo071014r
Return to citation in text: [1] -
Kusama, H.; Karibe, Y.; Onizawa, Y.; Iwasawa, N. Angew. Chem., Int. Ed. 2010, 49, 4269. doi:10.1002/anie.201001061
Return to citation in text: [1] -
Ito, H.; Ohmiya, H.; Sawamura, M. Org. Lett. 2010, 12, 4380. doi:10.1021/ol101860j
Return to citation in text: [1]
| 1. | Ciochina, R.; Grossman, R. B. Chem. Rev. 2006, 106, 3963. doi:10.1021/cr0500582 |
| 13. | Barriault, L.; Barabé, F.; Bétournay, G.; Bellavance, G. Org. Lett. 2009, 11, 4236. doi:10.1021/ol901722q |
| 9. | Shimizu, Y.; Shi, S. L.; Usuda, H.; Kanai, M.; Shibasaki, M. Angew. Chem., Int. Ed. 2010, 49, 1103. doi:10.1002/anie.200906678 |
| 10. | Kuramochi, A.; Usuda, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 14200. doi:10.1021/ja055301t |
| 11. | Danishefsky, S. J.; Siegel, D. R. J. Am. Chem. Soc. 2006, 128, 1048. doi:10.1021/ja057418n |
| 12. | Ahmad, N. M.; Rodeschini, V.; Simpkins, N. S.; Ward, S. E.; Blake, A. J. J. Org. Chem. 2007, 72, 4803. doi:10.1021/jo070388h |
| 7. | Fukuyama, Y.; Kuwayama, A.; Minami, H. Chem. Pharm. Bull. 1997, 45, 947. |
| 8. | Fukuyama, Y.; Minami, H.; Kuwayama, A. Phytochemistry 1998, 49, 853. doi:10.1016/S0031-9422(98)00126-5 |
| 2. | Bystrov, N. S.; Chernov, B. K.; Dobrynin, V. N.; Kolosov, M. N. Tetrahedron Lett. 1975, 16, 2791. doi:10.1016/S0040-4039(00)75241-5 |
| 3. | Müller, W. E.; Singer, A.; Wonnemann, M.; Hafner, U.; Rolli, M.; Schäfer, C. Pharmacopsychiatry 1998, 31, 16. doi:10.1055/s-2007-979341 |
| 4. | Verotta, L.; Appendino, G.; Bombardelli, E.; Brun, R. Bioorg. Med. Chem. Lett. 2007, 17, 1544. doi:10.1016/j.bmcl.2006.12.100 |
| 5. | Gey, C.; Kyrylenko, S.; Henning, L.; Nguyen, L.-H. D.; Büttner, A.; Pham, H. D.; Giannis, A. Angew. Chem., Int. Ed. 2007, 46, 5219. doi:10.1002/anie.200605207 |
| 6. | Moore, L. B.; Goodwin, B.; Jones, S. A.; Wisely, G. B.; Serabjit-Singh, C. J.; Willson, T. M.; Collins, J. L.; Kliewer, S. A. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7500. doi:10.1073/pnas.130155097 |
| 24. | Dankwardt, J. W. Tetrahedron Lett. 2001, 42, 5809. doi:10.1016/S0040-4039(01)01146-7 |
| 25. | Suhre, M. H.; Reif, M.; Kirsch, S. F. Org. Lett. 2005, 7, 3925. doi:10.1021/ol0514101 |
| 26. | Staben, S. T.; Kennedy-Smith, J. J.; Huang, D.; Corkey, B. K.; LaLonde, R. L.; Toste, F. D. Angew. Chem., Int. Ed. 2006, 45, 5991. doi:10.1002/anie.200602035 |
| 27. | Linghu, X.; Kennedy-Smith, J. J.; Toste, F. D. Angew. Chem., Int. Ed. 2007, 46, 7671. doi:10.1002/anie.200702695 |
| 28. | Lee, K.; Lee, P. H. Adv. Synth. Catal. 2007, 349, 2092. doi:10.1002/adsc.200700304 |
| 29. | Minnihan, E. C.; Colletti, S. L.; Toste, F. D.; Shen, H. C. J. Org. Chem. 2007, 72, 6287. doi:10.1021/jo071014r |
| 30. | Kusama, H.; Karibe, Y.; Onizawa, Y.; Iwasawa, N. Angew. Chem., Int. Ed. 2010, 49, 4269. doi:10.1002/anie.201001061 |
| 31. | Ito, H.; Ohmiya, H.; Sawamura, M. Org. Lett. 2010, 12, 4380. doi:10.1021/ol101860j |
| 23. | Johnson, W. S.; McCarry, B. E.; Markezich, R. L.; Boots, S. G. J. Am. Chem. Soc. 1980, 102, 352. doi:10.1021/ja00521a057 |
| 14. | Toste, F. D.; Gorin, D. J. Nature 2007, 446, 395. doi:10.1038/nature05592 |
| 15. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333. doi:10.1039/B612008C |
| 16. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335 |
| 17. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x |
| 18. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g |
| 19. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l |
| 20. | Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d |
| 21. | Skouta, R.; Li, C.-J. Tetrahedron 2008, 64, 4917. doi:10.1016/j.tet.2008.03.083 |
| 22. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 5, 675. doi:10.1055/s-0029-1219369 |
© 2011 Sow et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)