Abstract
A regioselective synthesis of 6-ω-alkenyluridines 3, precursors of potent antiviral and antitumor cyclonucleosides 5, is described. While ω-alkenyl halides do not alkylate 6-lithiouridine, compounds 3 were prepared in a regioselective manner by sequential treatment of 6-methyluridine 2 with LTMP or LDA (4 equiv) in THF at −30 °C followed by alkylation with ω-alkenyl bromides.

Graphical Abstract
Introduction
Conformationally restricted C–C bridged cyclonucleosides bearing a linkage between the sugar moiety and the nucleobase, exhibit a broad spectrum of antiviral and antitumor activities [1-4]. Cyclonucleosides are excellent tools for studying the role of the conformational parameters that are critical for the design of new nucleoside drug candidates [4-8]. These cyclic compounds are expected to have a beneficial biological impact especially toward enzymatic repair processes [9].
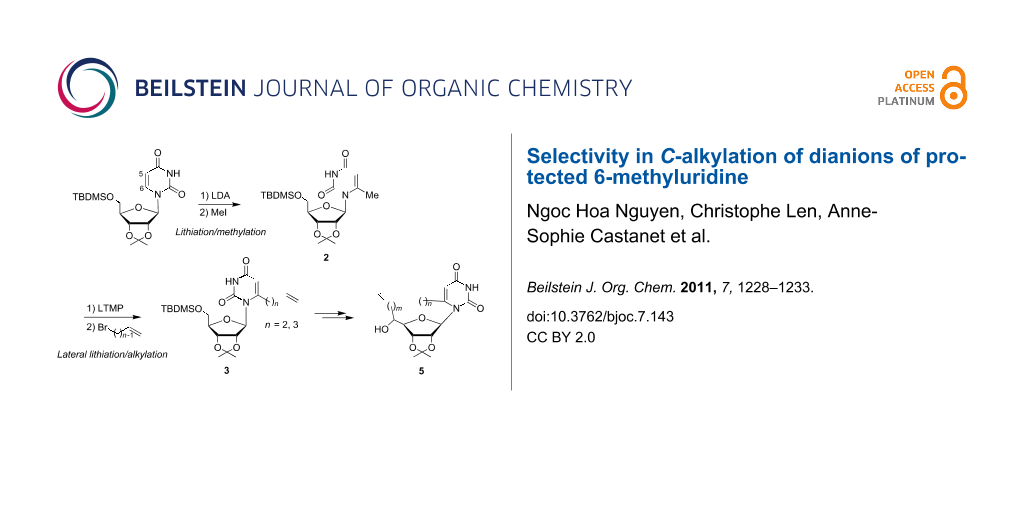
As part of an ongoing program directed by one of us (C. L.) toward the synthesis and development of new cyclonucleosides 5 [5,6], we envisioned that the general transformation outlined in Scheme 1 might afford a facile entry to 5 from dialkenyl precursors 4 by ring-closing metathesis [10-12]. The strategy relies on the preparation of unknown 6-ω-alkenyluridine key intermediates 3. We report herein that sequential ring lithiation/methylation of the simple protected uridine 1 leading to 2 followed by lateral lithiation/alkylation with ω-alkenyl bromides provides a useful regioselective chain-extension procedure and an efficient route to 3.
![[1860-5397-7-143-i1]](/bjoc/content/inline/1860-5397-7-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of potent antiviral and antitumor cyclonucleosides 5.
Scheme 1: Synthesis of potent antiviral and antitumor cyclonucleosides 5.
Results and Discussion
Most methods for the construction of C-substituted nucleosides are based on ring lithiation of nucleoside derivatives followed by their reaction with appropriate electrophiles. Thus, sequential lithiation of 2',3'-O-isopropylideneuridine (6) with LDA in THF (Figure 1) and electrophilic quenching with n-bromobutane was reported to give 6-n-butyl-2',3'-O-isopropylideneuridine (8) in a regiospecific manner (60%) [13]. It seems likely that the reaction proceeds via trianion 7 where the 5'-OLi group can easily participate in the stabilization of the 6-lithio intermediate. ω-Alkenyl bromides are known to be poor electrophiles toward organolithiums [14], and indeed, 7 failed to react, in our experiments, with 4-bromo-but-1-ene to give 9.
![[1860-5397-7-143-1]](/bjoc/content/figures/1860-5397-7-143-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Lithiation of 2',3'-O-isopropylideneuridine (6).
Figure 1: Lithiation of 2',3'-O-isopropylideneuridine (6).
We then turned our attention to the metalation of the 5'-O-TBDMS protected nucleoside 10 (Figure 2). Treatment with LDA (5 equiv) in THF at −70 °C followed by addition of D2O provided 12 in 82% yield (evaluated by NMR) with exclusive deuterium incorporation at the C6 position. However, almost complete recovery of the starting material was observed when dianion 11 was allowed to react with 4-bromobut-1-ene [15]. Lithium–copper transmetallation was also attempted. Unfortunately, addition of 0.25 equiv of Li2CuCl4 [16-18] to 11 followed by quenching with 4-bromobut-1-ene failed to produce 3a.
![[1860-5397-7-143-2]](/bjoc/content/figures/1860-5397-7-143-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Metalation of 5'-O-TMDMS protected nucleoside 10.
Figure 2: Metalation of 5'-O-TMDMS protected nucleoside 10.
Consequently, lateral lithiations were examined. Lateral lithiation of benzenoid aromatics requires a stabilizing group capable of either delocalizing negative charge or stabilizing an organolithium by coordination [19,20]. Primary, allylic, and benzylic halides usually give good yields of laterally alkylated products. Secondary and acetylenic halides have been used in several instances. Successful reaction with these substrates is noteworthy since many aryllithiums arising from ortho-lithiation reactions do not alkylate, or give poor yields, with any halides other than iodomethane [21-24]. Competing base-induced elimination reactions are presumably observed with iodoethane and higher homologues [19,20,25,26]. It has also been proposed that poor reactivity of lithiated carbanions toward alkyl halides may result from steric hindrance [24,27]. Recently, the lateral lithiation of 4-hydroxy-6,7-dimethoxy-8-methyl-2-naphthoic acid was applied to the regioselective efficient construction of a series of 5,5'-didesisopropyl-5,5'-dialkylapogossypol derivatives that are potent pan-active inhibitors of anti-apoptotic Bcl-2 family proteins [28].
Literature furnishes little information regarding lateral lithiations in the nucleoside field and the data, scarce as they are, even appear to be inconsistent at first sight. Treatment of 2',3',5'-tri-O-benzoyl-3,6-dimethyluridine (13) with chloroacetone or 2-chloroacetophenone in the presence of LDA (1.2 equiv, THF, −78 °C) afforded 6-(oxiranylmethyl)uridine derivatives 14 exclusively (Figure 3) [29]. With 5-chloro-2-pentanone, the reaction led to a mixture of 5- and 6-substituted uridine regioisomers 15 and 16 in 47% and 28% yield, respectively. It was suggested that the N-1 sugar moiety in the syn orientation of the nucleoside might affect the access of a very sterically demanding electrophile, such as 5-chloro-2-pentanone, to the 6-position. This hypothesis was confirmed by a probe experiment where an even more sterically hindered racemic 3-bromocamphor was used as an electrophile. The corresponding C5-alkylated uridine derivative was obtained as the only recovered product, in low yield (23%), besides the unreacted substrate.
![[1860-5397-7-143-3]](/bjoc/content/figures/1860-5397-7-143-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Lithiation/alkylation of 2',3',5'-tri-O-benzoyl-3,6-dimethyluridine (13) using LDA.
Figure 3: Lithiation/alkylation of 2',3',5'-tri-O-benzoyl-3,6-dimethyluridine (13) using LDA.
Having these precedents in mind, we decided to investigate the preparation of 6-ω-alkenyluridines 3 by lithiation of 6-methyluridine derivative 2. Miyasaka et al. observed concomitant formation of 6-ethyl derivative alongside the expected 6-methyl derivative when 2',3'-O-isopropylideneuridine (6) was allowed to react with LDA and treated with MeI [13]. We found similarly that 10 in the presence of LDA (2.5 equiv) followed by addition of MeI (3.3 equiv) at −78 °C gave a mixture of 6-methyluridine 2 (44%) and 6-ethyluridine 17 (17%) (Scheme 2). By slow addition of the preformed dianion 11 to a THF solution of MeI (reverse-addition mode) [30,31], 2 was produced in satisfactory yield (72%) while formation of 6-ethyluridine 17 was reduced to <5%. Apparently LDA does not coordinate with the substrate in this transformation [32]. 6-Methyluridine can also be synthesized from 5'-O-(tert-butyl-dimethylsilyl)-6-iodo-2',3'-O-isopropylideneuridine via palladium-catalyzed cross-coupling with Me4Sn [33].
![[1860-5397-7-143-i2]](/bjoc/content/inline/1860-5397-7-143-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of 2',3'-O-isopropylidene-5'-O-(tert-butyldimethylsilyl)-6-methyluridine (2).
Scheme 2: Preparation of 2',3'-O-isopropylidene-5'-O-(tert-butyldimethylsilyl)-6-methyluridine (2).
We have then embarked on a detailed investigation of the lithiation/alkylation of 6-methyluridine 2, varying the base, metalation temperature, and exposure times (Scheme 3). We were concerned with the question of relative acidity of the methyl (C7) and the C5 centers that can compete through 18A or 18B [34-36]. In fact the two negative charges at N3 and C5/C7 are delocalized through the O2-C2-N3-C4-O4-C5-C6-C7 bond system rather than being localized dianions: All these forms are resonance structures of the same extended dianionic enolate 18 [37].
![[1860-5397-7-143-i3]](/bjoc/content/inline/1860-5397-7-143-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Lateral lithiation/alkylation of 6-methyluridine 2.
Scheme 3: Lateral lithiation/alkylation of 6-methyluridine 2.
In the past, the regioselectivity of reactions of allyl anions have sometimes been explained using the HSAB theory [34]. In the present case, soft electrophiles (ω-alkenyl bromides) are used in the alkylation reaction. However, it is not straightforward to predict the softest center of 18. In the literature, the regioselectivity of lithiation of allyl anions substituted by one nitrogen at the central carbon (C=C(N)–C) has scarcely been studied [38-40]. Deprotonation of simple enamines or allylamines employing n-BuLi and t-BuLi/t-BuOK produced nitrogen-substituted allylic anions which undergo protonation, alkylation, trimethylsilylation and reaction with carbonyl compounds and epoxides either exclusively or predominantly at the γ-position [41-43]. Previous work also showed that cyclic enaminoketones, esters and nitriles were converted into their enolate with n-BuLi and alkylated with a variety of alkylating agents, affording the product of an exclusive γ-alkylation [42-45].
The results are summarized in Table 1. An excess of LDA (4 equiv) at −70 °C produced a dilithium reagent, which was presumed to be 18, as a yellow solution. The colour faded when allyl bromide (8 equiv/ −70 °C → rt/12 h) was introduced, providing a mixture of regioisomers 3a and 19a which were separated by chromatography (entry 1, 58% and 10% yield, respectively). With LTMP, the delivery of a less acidic conjugated amine (TMP) in the reaction medium could be expected to prevent protonation of lithiated intermediates and thus to allow more efficient trapping by an electrophile [46,47]. Indeed, LTMP gave a better yield, but a slight decrease in the regioselectivity was observed (3a/19a 65:20). The 1H NMR spectrum of 19a displayed a singlet at 2.31 ppm, attributed to the C7-methyl group, and the disappearance of the H5 signal at 5.57 ppm. 4-Bromobut-1-ene underwent exclusive C7-carbanion alkylation to 3b in good yield, with no indication of products resulting from C5-alkylation (entries 3 and 4).
Allyl bromide is a good electrophile that can react with both mesomeric forms 18A and 18B to give 3a and 19a. Apparently dianion 18B is not nucleophilic enough to react with 4-bromobut-1-ene to give 19b, and 3b is formed exclusively. The lateral alkylation of uridine enolate 18 was best accomplished through use of LDA or LTMP as the carbanion generating species, rather than LiHMDS or s-BuLi/TMEDA. The bis-allylated products 20 and 21 (Figure 4) were obtained in 40% and 18% yield, respectively with LiHMDS at −70 °C and quenched with allyl bromide (entry 5). This result suggests the remetalation of 3a is faster than the destruction of LiHMDS by the excess of allyl bromide. Structure of 20 was confirmed by 1H NMR and a two-dimensional COSY experiment, which allowed the assignment of the proton–proton correlations of H7 and the allylic methylene groups. Metalation with s-BuLi/TMEDA complex was less efficient although the reaction did not lead to degradation products (entry 6). Ring/internal lithiations of uridine derivatives with s-BuLi/TMEDA are usually performed with fully TBDMS-protected ribofuranose nucleosides to allow better regiochemical control and to prevent nucleophilic attack of the base on the sugar moiety [48].
![[1860-5397-7-143-4]](/bjoc/content/figures/1860-5397-7-143-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Bis-allylated products 20 and 21.
Figure 4: Bis-allylated products 20 and 21.
Conclusion
In summary, a straightforward approach to 6-ω-alkenyluridines 3 from readily available protected uridine 1 is proposed. Whereas direct ring alkylation of 6-lithiated uridine 11 with ω-alkenyl bromides failed, our approach relies on lateral lithiation/alkylation of 6-methyluridine 2. The total synthesis and biological properties of cyclonucleosides 5 will be reported separately.
Supporting Information
| Supporting Information File 1: Experimental section (preparation and spectral data of compounds). | ||
| Format: PDF | Size: 78.7 KB | Download |
References
-
Len, C.; Mondon, M.; Lebreton, J. Tetrahedron 2008, 64, 7453–7475. doi:10.1016/j.tet.2008.04.095
Return to citation in text: [1] -
Mieczkowski, A.; Roy, V.; Agrofoglio, L. A. Chem. Rev. 2010, 110, 1828–1856. doi:10.1021/cr900329y
Return to citation in text: [1] -
Mieczkowski, A.; Agrofoglio, L. A. Curr. Med. Chem. 2010, 17, 1527–1549. doi:10.2174/092986710790979962
Return to citation in text: [1] -
Chu, C. K., Ed. Antiviral Nucleosides: Chiral Synthesis and Chemotherapy, 1st ed.; Elsevier: Amsterdam, 2003.
Return to citation in text: [1] [2] -
Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron 2008, 64, 7828–7836. doi:10.1016/j.tet.2008.05.112
Return to citation in text: [1] [2] -
Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron Lett. 2006, 47, 6221–6224. doi:10.1016/j.tetlet.2006.06.145
Return to citation in text: [1] [2] -
Viña, D.; Quezada, E.; Santana, L.; Uriarte, E. Tetrahedron 2006, 62, 9949–9952. doi:10.1016/j.tet.2006.08.007
Return to citation in text: [1] -
Yoshimura, Y.; Yamazaki, Y.; Wachi, K.; Satoh, S.; Takahata, H. Synlett 2007, 111–114. doi:10.1055/s-2006-956464
Return to citation in text: [1] -
Muller, E.; Gasparutto, D.; Jaquinod, M.; Romieu, A.; Cadet, J. Tetrahedron 2000, 56, 8689–8701. doi:10.1016/S0040-4020(00)00809-7
Return to citation in text: [1] -
Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1–3.
Return to citation in text: [1] -
Amblard, F.; Nolan, S. P.; Agrofoglio, L. A. Tetrahedron 2005, 61, 7067–7080. doi:10.1016/j.tet.2005.04.040
Return to citation in text: [1] -
Agrofoglio, L. A.; Nolan, S. P. Curr. Top. Med. Chem. 2005, 5, 1541–1558. doi:10.2174/156802605775009739
Return to citation in text: [1] -
Tanaka, H.; Nasu, I.; Miyasaka, T. Tetrahedron Lett. 1979, 20, 4755–4758. doi:10.1016/S0040-4039(01)86702-2
Return to citation in text: [1] [2] -
Knight, D. W. Alkylations of Vinyl Carbanions. In Carbon–Carbon σ-Bond Formation; Trost, B. M.; Fleming, I., Eds.; Comprehensive Organic Synthesis, Vol. 3; Pergamon: Oxford, 1991; pp 241–270. doi:10.1016/B978-0-08-052349-1.00063-9
Return to citation in text: [1] -
Mieczkowski, A.; Peltier, P.; Zevaco, T.; Agrofoglio, L. A. Tetrahedron 2009, 65, 4053–4059.
Return to citation in text: [1] -
Gelin, J.; Mortier, J.; Moyroud, J.; Chêne, A. J. Org. Chem. 1993, 58, 3473–3475. doi:10.1021/jo00065a001
Return to citation in text: [1] -
Moyroud, J.; Gelin, J.; Chêne, A.; Mortier, J. Tetrahedron 1996, 52, 8525–8534. doi:10.1016/0040-4020(96)00408-5
Return to citation in text: [1] -
Berkheij, M.; van der Sluis, L.; Sewing, C.; den Boer, D. J.; Terpstra, J. W.; Hiemstra, H.; Bakker, W. I. I.; van den Hoogenband, A.; van Maarseveen, J. H. Tetrahedron Lett. 2005, 46, 2369–2371. doi:10.1016/j.tetlet.2005.02.085
Return to citation in text: [1] -
Clark, R. D.; Jahangir, A. Org. React. 1995, 47, 1–314. doi:10.1002/0471264180.or047.01
Return to citation in text: [1] [2] -
Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon: Oxford, 2002; pp 73–85.
Return to citation in text: [1] [2] -
Bennetau, B.; Mortier, J.; Moyroud, J.; Guesnet, J.-L. J. Chem. Soc., Perkin Trans. 1 1995, 1265–1271. doi:10.1039/P19950001265
Return to citation in text: [1] -
Flippin, L. A. Tetrahedron Lett. 1991, 32, 6857–6860. doi:10.1016/0040-4039(91)80425-6
Return to citation in text: [1] -
Buttery, C. D.; Knight, D. W.; Nott, A. P. Tetrahedron Lett. 1982, 23, 4127–4130. doi:10.1016/S0040-4039(00)88366-5
Return to citation in text: [1] -
Comins, D. L.; Brown, J. D.; Mantlo, N. B. Tetrahedron Lett. 1982, 23, 3979–3982. doi:10.1016/S0040-4039(00)88674-8
Return to citation in text: [1] [2] -
Carruthers, W.; Coldham, I. Modern Methods of Organic Synthesis, 4th ed.; Cambridge University Press: Cambridge, 2004; p 61.
Return to citation in text: [1] -
Tarnchompoo, B.; Thebtaranonth, C.; Thebtaranonth, Y. Tetrahedron Lett. 1990, 31, 5779–5780. doi:10.1016/S0040-4039(00)97957-7
Return to citation in text: [1] -
Harris, T. D.; Roth, G. P. J. Org. Chem. 1979, 44, 2004–2007. doi:10.1021/jo01326a024
Return to citation in text: [1] -
Le, T. T.; Chau, N. T. T.; Nguyen, T. T.; Brien, J.; Thai, T. T.; Nourry, A.; Castanet, A.-S.; Nguyen, K. P. P.; Mortier, J. J. Org. Chem. 2011, 76, 601–608. doi:10.1021/jo102147s
Return to citation in text: [1] -
Saladino, R.; Crestini, C.; Palamara, A. T.; Danti, M. C.; Manetti, F.; Corelli, F.; Garaci, E.; Botta, M. J. Med. Chem. 2001, 44, 4554–4562.
Return to citation in text: [1] -
Mortier, J.; Vaultier, M.; Cantegril, R.; Dellis, P. Aldrichimica Acta 1997, 30, 34.
Return to citation in text: [1] -
Tanaka, H.; Hayakawa, H.; Miyasaka, T. Lithiation Chemistry of Uridine Derivatives: Access to a New Anti-HIV-1 Lead. In Nucleosides and Nucleotides As Antitumor and Antiviral Agents, Proceedings of American Chemical Society Carbohydrate Division Symposium, San Francisco, California, April 7, 1992; Baker, D. C.; Chu, C. K., Eds.; 1992; pp 23–54.
Return to citation in text: [1] -
Marsais, F.; Cronnier, A.; Trecourt, F.; Queguiner, G. J. Org. Chem. 1987, 52, 1133–1136. doi:10.1021/jo00382a029
Return to citation in text: [1] -
Tanaka, H.; Hayakawa, H.; Shibata, S.; Haraguchi, K.; Miyasaka, T.; Hirota, K. Nucleosides, Nucleotides Nucleic Acids 1992, 11, 319–328. doi:10.1080/07328319208021706
Return to citation in text: [1] -
Katrizky, A. R.; Piffl, M.; Lang, H.; Anders, E. Chem. Rev. 1999, 99, 665–722. doi:10.1021/cr9404297
Return to citation in text: [1] [2] -
Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207–2293. doi:10.1021/cr00022a010
Return to citation in text: [1] -
Stowell, J. C. Chem. Rev. 1984, 84, 409–435. doi:10.1021/cr00063a001
Return to citation in text: [1] -
Kwiatkowski, J. S.; Pullman, B. Tautomerism and Electronic Structure of Biological Pyrimidines. In Advances in Heterocyclic Chemistry; Katritzki, A. R.; Boulton, A. J., Eds.; Academic Press: New York, 1975; Vol. 18, pp 199–335. doi:10.1016/S0065-2725(08)60131-2
Return to citation in text: [1] -
Cimarelli, C.; Palmieri, G.; Camalli, M. Tetrahedron 1997, 53, 6893–6902. doi:10.1016/S0040-4020(97)00320-7
Return to citation in text: [1] -
Bartoli, G.; Bosco, M.; Cimarelli, C.; Dalpozzo, R.; Guerra, M.; Palmieri, G. J. Chem. Soc., Perkin Trans. 2 1992, 649–650. doi:10.1039/p29920000649
Return to citation in text: [1] -
Hu, T. Q.; Weiler, L. Can. J. Chem. 1994, 72, 1500–1511. doi:10.1139/v94-187
Return to citation in text: [1] -
Ahlbrecht, H.; Sudheendranath, C. S. Synthesis 1982, 717–719. doi:10.1055/s-1982-29910
Return to citation in text: [1] -
Ahlbrecht, H.; Simon, H. Synthesis 1983, 58–60. doi:10.1055/s-1983-30223
Return to citation in text: [1] [2] -
Costisella, B.; Gross, H. Tetrahedron 1982, 38, 139–145. doi:10.1016/0040-4020(82)85057-6
Return to citation in text: [1] [2] -
Bryson, T. A.; Gammill, R. B. Tetrahedron Lett. 1974, 15, 3963–3966. doi:10.1016/S0040-4039(01)92057-X
Return to citation in text: [1] -
Chen, Y. L.; Mariano, P. S.; Little, G. M.; O'Brien, D.; Huesmann, P. L. J. Org. Chem. 1981, 46, 4643–4654. doi:10.1021/jo00336a002
Return to citation in text: [1] -
Fraser, R. R.; Mansour, T. S. J. Org. Chem. 1984, 49, 3442–3443.
Return to citation in text: [1] -
Gros, P.; Choppin, S.; Fort, Y. J. Org. Chem. 2003, 68, 2243–2247. doi:10.1021/jo026559u
Return to citation in text: [1] -
Hayakawa, H.; Tanaka, H.; Obi, K.; Itoh, M.; Miyasaka, T. Tetrahedron Lett. 1987, 28, 87–90. doi:10.1016/S0040-4039(00)95656-9
Return to citation in text: [1]
| 1. | Len, C.; Mondon, M.; Lebreton, J. Tetrahedron 2008, 64, 7453–7475. doi:10.1016/j.tet.2008.04.095 |
| 2. | Mieczkowski, A.; Roy, V.; Agrofoglio, L. A. Chem. Rev. 2010, 110, 1828–1856. doi:10.1021/cr900329y |
| 3. | Mieczkowski, A.; Agrofoglio, L. A. Curr. Med. Chem. 2010, 17, 1527–1549. doi:10.2174/092986710790979962 |
| 4. | Chu, C. K., Ed. Antiviral Nucleosides: Chiral Synthesis and Chemotherapy, 1st ed.; Elsevier: Amsterdam, 2003. |
| 10. | Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1–3. |
| 11. | Amblard, F.; Nolan, S. P.; Agrofoglio, L. A. Tetrahedron 2005, 61, 7067–7080. doi:10.1016/j.tet.2005.04.040 |
| 12. | Agrofoglio, L. A.; Nolan, S. P. Curr. Top. Med. Chem. 2005, 5, 1541–1558. doi:10.2174/156802605775009739 |
| 29. | Saladino, R.; Crestini, C.; Palamara, A. T.; Danti, M. C.; Manetti, F.; Corelli, F.; Garaci, E.; Botta, M. J. Med. Chem. 2001, 44, 4554–4562. |
| 5. | Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron 2008, 64, 7828–7836. doi:10.1016/j.tet.2008.05.112 |
| 6. | Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron Lett. 2006, 47, 6221–6224. doi:10.1016/j.tetlet.2006.06.145 |
| 13. | Tanaka, H.; Nasu, I.; Miyasaka, T. Tetrahedron Lett. 1979, 20, 4755–4758. doi:10.1016/S0040-4039(01)86702-2 |
| 9. | Muller, E.; Gasparutto, D.; Jaquinod, M.; Romieu, A.; Cadet, J. Tetrahedron 2000, 56, 8689–8701. doi:10.1016/S0040-4020(00)00809-7 |
| 24. | Comins, D. L.; Brown, J. D.; Mantlo, N. B. Tetrahedron Lett. 1982, 23, 3979–3982. doi:10.1016/S0040-4039(00)88674-8 |
| 27. | Harris, T. D.; Roth, G. P. J. Org. Chem. 1979, 44, 2004–2007. doi:10.1021/jo01326a024 |
| 4. | Chu, C. K., Ed. Antiviral Nucleosides: Chiral Synthesis and Chemotherapy, 1st ed.; Elsevier: Amsterdam, 2003. |
| 5. | Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron 2008, 64, 7828–7836. doi:10.1016/j.tet.2008.05.112 |
| 6. | Zhong, S.; Mondon, M.; Pilard, S.; Len, C. Tetrahedron Lett. 2006, 47, 6221–6224. doi:10.1016/j.tetlet.2006.06.145 |
| 7. | Viña, D.; Quezada, E.; Santana, L.; Uriarte, E. Tetrahedron 2006, 62, 9949–9952. doi:10.1016/j.tet.2006.08.007 |
| 8. | Yoshimura, Y.; Yamazaki, Y.; Wachi, K.; Satoh, S.; Takahata, H. Synlett 2007, 111–114. doi:10.1055/s-2006-956464 |
| 28. | Le, T. T.; Chau, N. T. T.; Nguyen, T. T.; Brien, J.; Thai, T. T.; Nourry, A.; Castanet, A.-S.; Nguyen, K. P. P.; Mortier, J. J. Org. Chem. 2011, 76, 601–608. doi:10.1021/jo102147s |
| 16. | Gelin, J.; Mortier, J.; Moyroud, J.; Chêne, A. J. Org. Chem. 1993, 58, 3473–3475. doi:10.1021/jo00065a001 |
| 17. | Moyroud, J.; Gelin, J.; Chêne, A.; Mortier, J. Tetrahedron 1996, 52, 8525–8534. doi:10.1016/0040-4020(96)00408-5 |
| 18. | Berkheij, M.; van der Sluis, L.; Sewing, C.; den Boer, D. J.; Terpstra, J. W.; Hiemstra, H.; Bakker, W. I. I.; van den Hoogenband, A.; van Maarseveen, J. H. Tetrahedron Lett. 2005, 46, 2369–2371. doi:10.1016/j.tetlet.2005.02.085 |
| 21. | Bennetau, B.; Mortier, J.; Moyroud, J.; Guesnet, J.-L. J. Chem. Soc., Perkin Trans. 1 1995, 1265–1271. doi:10.1039/P19950001265 |
| 22. | Flippin, L. A. Tetrahedron Lett. 1991, 32, 6857–6860. doi:10.1016/0040-4039(91)80425-6 |
| 23. | Buttery, C. D.; Knight, D. W.; Nott, A. P. Tetrahedron Lett. 1982, 23, 4127–4130. doi:10.1016/S0040-4039(00)88366-5 |
| 24. | Comins, D. L.; Brown, J. D.; Mantlo, N. B. Tetrahedron Lett. 1982, 23, 3979–3982. doi:10.1016/S0040-4039(00)88674-8 |
| 15. | Mieczkowski, A.; Peltier, P.; Zevaco, T.; Agrofoglio, L. A. Tetrahedron 2009, 65, 4053–4059. |
| 19. | Clark, R. D.; Jahangir, A. Org. React. 1995, 47, 1–314. doi:10.1002/0471264180.or047.01 |
| 20. | Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon: Oxford, 2002; pp 73–85. |
| 25. | Carruthers, W.; Coldham, I. Modern Methods of Organic Synthesis, 4th ed.; Cambridge University Press: Cambridge, 2004; p 61. |
| 26. | Tarnchompoo, B.; Thebtaranonth, C.; Thebtaranonth, Y. Tetrahedron Lett. 1990, 31, 5779–5780. doi:10.1016/S0040-4039(00)97957-7 |
| 14. | Knight, D. W. Alkylations of Vinyl Carbanions. In Carbon–Carbon σ-Bond Formation; Trost, B. M.; Fleming, I., Eds.; Comprehensive Organic Synthesis, Vol. 3; Pergamon: Oxford, 1991; pp 241–270. doi:10.1016/B978-0-08-052349-1.00063-9 |
| 13. | Tanaka, H.; Nasu, I.; Miyasaka, T. Tetrahedron Lett. 1979, 20, 4755–4758. doi:10.1016/S0040-4039(01)86702-2 |
| 19. | Clark, R. D.; Jahangir, A. Org. React. 1995, 47, 1–314. doi:10.1002/0471264180.or047.01 |
| 20. | Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon: Oxford, 2002; pp 73–85. |
| 33. | Tanaka, H.; Hayakawa, H.; Shibata, S.; Haraguchi, K.; Miyasaka, T.; Hirota, K. Nucleosides, Nucleotides Nucleic Acids 1992, 11, 319–328. doi:10.1080/07328319208021706 |
| 30. | Mortier, J.; Vaultier, M.; Cantegril, R.; Dellis, P. Aldrichimica Acta 1997, 30, 34. |
| 31. | Tanaka, H.; Hayakawa, H.; Miyasaka, T. Lithiation Chemistry of Uridine Derivatives: Access to a New Anti-HIV-1 Lead. In Nucleosides and Nucleotides As Antitumor and Antiviral Agents, Proceedings of American Chemical Society Carbohydrate Division Symposium, San Francisco, California, April 7, 1992; Baker, D. C.; Chu, C. K., Eds.; 1992; pp 23–54. |
| 32. | Marsais, F.; Cronnier, A.; Trecourt, F.; Queguiner, G. J. Org. Chem. 1987, 52, 1133–1136. doi:10.1021/jo00382a029 |
| 46. | Fraser, R. R.; Mansour, T. S. J. Org. Chem. 1984, 49, 3442–3443. |
| 47. | Gros, P.; Choppin, S.; Fort, Y. J. Org. Chem. 2003, 68, 2243–2247. doi:10.1021/jo026559u |
| 48. | Hayakawa, H.; Tanaka, H.; Obi, K.; Itoh, M.; Miyasaka, T. Tetrahedron Lett. 1987, 28, 87–90. doi:10.1016/S0040-4039(00)95656-9 |
| 41. | Ahlbrecht, H.; Sudheendranath, C. S. Synthesis 1982, 717–719. doi:10.1055/s-1982-29910 |
| 42. | Ahlbrecht, H.; Simon, H. Synthesis 1983, 58–60. doi:10.1055/s-1983-30223 |
| 43. | Costisella, B.; Gross, H. Tetrahedron 1982, 38, 139–145. doi:10.1016/0040-4020(82)85057-6 |
| 42. | Ahlbrecht, H.; Simon, H. Synthesis 1983, 58–60. doi:10.1055/s-1983-30223 |
| 43. | Costisella, B.; Gross, H. Tetrahedron 1982, 38, 139–145. doi:10.1016/0040-4020(82)85057-6 |
| 44. | Bryson, T. A.; Gammill, R. B. Tetrahedron Lett. 1974, 15, 3963–3966. doi:10.1016/S0040-4039(01)92057-X |
| 45. | Chen, Y. L.; Mariano, P. S.; Little, G. M.; O'Brien, D.; Huesmann, P. L. J. Org. Chem. 1981, 46, 4643–4654. doi:10.1021/jo00336a002 |
| 34. | Katrizky, A. R.; Piffl, M.; Lang, H.; Anders, E. Chem. Rev. 1999, 99, 665–722. doi:10.1021/cr9404297 |
| 38. | Cimarelli, C.; Palmieri, G.; Camalli, M. Tetrahedron 1997, 53, 6893–6902. doi:10.1016/S0040-4020(97)00320-7 |
| 39. | Bartoli, G.; Bosco, M.; Cimarelli, C.; Dalpozzo, R.; Guerra, M.; Palmieri, G. J. Chem. Soc., Perkin Trans. 2 1992, 649–650. doi:10.1039/p29920000649 |
| 40. | Hu, T. Q.; Weiler, L. Can. J. Chem. 1994, 72, 1500–1511. doi:10.1139/v94-187 |
| 34. | Katrizky, A. R.; Piffl, M.; Lang, H.; Anders, E. Chem. Rev. 1999, 99, 665–722. doi:10.1021/cr9404297 |
| 35. | Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207–2293. doi:10.1021/cr00022a010 |
| 36. | Stowell, J. C. Chem. Rev. 1984, 84, 409–435. doi:10.1021/cr00063a001 |
| 37. | Kwiatkowski, J. S.; Pullman, B. Tautomerism and Electronic Structure of Biological Pyrimidines. In Advances in Heterocyclic Chemistry; Katritzki, A. R.; Boulton, A. J., Eds.; Academic Press: New York, 1975; Vol. 18, pp 199–335. doi:10.1016/S0065-2725(08)60131-2 |
© 2011 Nguyen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)