Abstract
A new two-step general approach to [2.2]metacyclophane synthesis from substituted m-xylenes is described. The strategy employs a selective benzylic metalation and oxidative C–C bond formation for both synthetic operations. Regioselective benzylic metalation is achieved using the BuLi, KOt-Bu, TMP(H) (2,2,6,6-tetramethylpiperidine) combination (LiNK metalation conditions) and oxidative coupling with 1,2-dibromoethane. The synthetic ease of this approach compares favourably with previously reported methods and allows for ready access to potentially useful planar chiral derivatives.

Graphical Abstract
Introduction
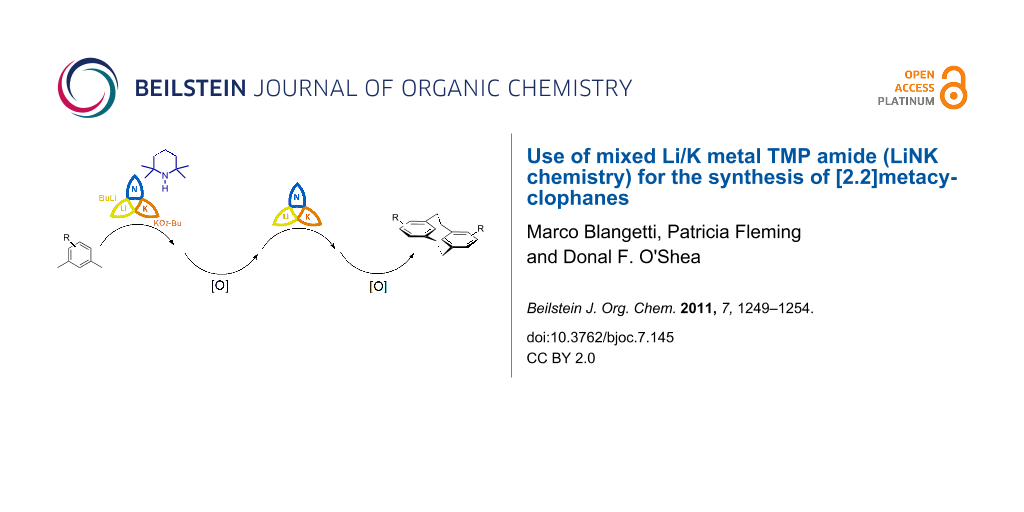
While direct metalation reactions are an essential contribution to the repertoire of modern synthetic methods, an underlying and often underestimated challenge remains in the achievement of predictable selective metalations of substrates that offer several potential sites of reaction. Examples of such challenges include the selective aryl metalation of arenes containing more than one directing group (DG), arene metalation in positions not ortho to the directing group, or the identification of reaction conditions to achieve selective benzylic metalation of substituted toluenes 1 to provide 3 (Scheme 1) [1-5]. We recently reported that mixed Li/K metal TMP amide (LiNK metalation conditions) is uniquely suited for the selective achievement of challenging metalations. Specifically, the use of the reagent triad BuLi/KOt-Bu/TMP(H) to generate a mixed Li/K metal TMP amide in situ has proven to be an efficient and general method to achieve vinyl and benzylic metalations with excellent selectivity [6,7]. We now exploit this selective benzylic metalation protocol for the specific synthesis of [2.2]metacyclophanes.
![[1860-5397-7-145-i1]](/bjoc/content/inline/1860-5397-7-145-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Selective benzylic metalation with LiNK conditions. DG = directing group.
Scheme 1: Selective benzylic metalation with LiNK conditions. DG = directing group.
Chemists have a long-standing fascination for the [2.2]cyclophane structures and have extensively studied their unusual physical and chemical properties induced by the close spatial proximity of their aryl rings [8]. Undoubtedly the most studied of the series (ortho, meta and para) are the [2.2]paracyclophanes, which have seen a recent resurgence of interest notably as planar chiral scaffolds for asymmetric catalysis [9-13]. Surprisingly, the [2.2]metacyclophanes, which also have the potential to be exploited as planar chiral templates, have received scant attention since the seminal reports of Schlögl in the early 1970s [14-16]. One possible explanation for this is the cumbersome methods required for their synthesis. Typical approaches have utilised Wurtz coupling, the oxidation and thermal (500–600 °C) extrusion of SO2 from dithia[3.3]cyclophanes or the photochemical ring contraction of diselena[3.3]cyclophanes [17-21].
We envisaged that our LiNK metalation conditions with in situ oxidative coupling could offer a facile general approach to [2.2]metacyclophanes, which would be of general synthetic interest (Scheme 2). Oxidative homo-coupling of benzyl anions has previously been noted, but it has remained relatively unexplored as a synthetic procedure [22-24]. We speculated that if oxidative coupling of the benzyl metalated xylenes 5 could be achieved to form the open dimer 6, then a second metalation and oxidative ring closure would yield the [2.2]metacyclophanes 8. A stepwise approach, as shown in Scheme 2, could allow for the introduction of different groups on each of the aryl rings. In addition, it could facilitate the synthesis of planar chiral derivatives without the complication of mixtures with achiral isomers being generated, which occurs if, for example, a 1-substituted-2,4-bis(halomethyl)benzene is used as the starting compound [15].
![[1860-5397-7-145-i2]](/bjoc/content/inline/1860-5397-7-145-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Iterative LiNK/oxidative coupling synthesis of [2.2]metacyclophanes.
Scheme 2: Iterative LiNK/oxidative coupling synthesis of [2.2]metacyclophanes.
Results and Discussion
In order to examine the scope and potential of this approach, five differently substituted xylenes were investigated, namely m-xylene (4a), mesitylene (4b), 1-methoxy-3,5-dimethylbenzene (4c), (3,5-dimethylphenyl)dimethylamine (4d) and 2,4-dimethylbenzoic acid (4e) (Figure 1). These xylenes present interesting challenges for metalation selectivity in that for derivatives 4a–d both methyl groups are equivalent and so the alternative metalation sites are in the aryl ring, whereas the challenge is elevated for 4e as it contains two differing benzylic sites and an ortho-aryl position primed for metalation by a strong directing group.
![[1860-5397-7-145-1]](/bjoc/content/figures/1860-5397-7-145-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Sequential treatment of 4a–d with BuLi, KOt-Bu and TMP(H) at −78 °C followed by the oxidant 1,2-dibromoethane [25] gave good to excellent yields of the targeted homo-dimer products 6a–d (Table 1, entries 1–4). It was also possible to form a “mixed dimer” by the reaction of 4a and 4c together, which gave 6e, containing one m-OCH3 substituted aryl ring, in a 22% yield (Table 1, entry 5) following chromatography to remove the other homo-coupled products (6a, 6c). A similar approach was used in a combined reaction of 4a and 4e, giving purified 4-methyl-2-(3-methylphenethyl)benzoic acid (6f) (Table 1, entry 6). This product is the result of oxidative hetero coupling of benzylic metalated xylene and 2-(methyl-metalated)-4-methylbenzoate.
Table 1: Oxidative coupling of benzylic metalated xylenes 4.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-145-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | R | Product | Yield % |
|---|---|---|---|---|
| 1 | 4a | H | 6a | 91 |
| 2 | 4b | CH3 | 6b | 72 |
| 3 | 4c | OCH3 | 6c | 92 |
| 4 | 4d | N(CH3)2 | 6d | 49 |
| 5 | 4a/4c | OCH3/H | 6e | 22a |
| 6 | 4a/4e | CO2H/H | 6f | 13b |
a6a and 6c also obtained in 14% and 49% yields respectively.
b6a and dimer of 4e also obtained in 11% and 62% yields respectively.
To more clearly illustrate a site selective metalation of 4e for the methyl group ortho to the carboxylate, this substrate was metalated using LiNK metalation conditions (using an addition equivalent of BuLi to first deprotonate the carboxylic acid) and quenched with CD3OD. It was anticipated that under our low temperature yet thermodynamically controlled conditions the selective site of metalation should be the more acidic 2-methyl position [26]. This was confirmed by 2H NMR, which showed that incorporated deuterium was above 90% in the 2-methyl position with less than 10% in the 4-methyl group and no detectable aryl deuteration (Figure 2).
![[1860-5397-7-145-2]](/bjoc/content/figures/1860-5397-7-145-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Metalation selectivity for 4e (arrows indicate potential metalation sites). 2H NMR spectrum in CH2Cl2. *CD2Cl2. **2-Methyl-4-D-methylbenzoic acid.
Figure 2: Metalation selectivity for 4e (arrows indicate potential metalation sites). 2H NMR spectrum in CH2Cl...
A similar experiment was carried out for the even more complex hetero-dimer substrate 6f, in which a selective di-benzylic metalation of the two different methyl positions was attempted. Treatment of 6f with three equivalents of BuLi (one to deprotonate the carboxylic acid) and two equivalents of KOt-Bu/TMP(H) followed by deuteration gave the di-deuterated product D2-6f. 2H NMR analysis showed no aryl or bridging methylene deuteration, with deuterium incorporated only into the two non-equivalent benzylic methyl positions (Figure 3).
![[1860-5397-7-145-3]](/bjoc/content/figures/1860-5397-7-145-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Di-metalation selectivity for 6f. 2H NMR spectrum in CH2Cl2. *CD2Cl2.
Figure 3: Di-metalation selectivity for 6f. 2H NMR spectrum in CH2Cl2. *CD2Cl2.
With the benzylic metalation confirmed, the second step to complete the [2.2]metacyclophane synthesis required identical conditions to the first to provide the di-benzylic metalated derivatives 7 (Table 2). We anticipated that an intramolecular ring closing by oxidative coupling would provide the desired cyclophane product in addition to open chain oligomers or larger ring systems.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-145-i4.svg?max-width=637&scale=1.0)
Substrates 6a–e were treated with two equivalents of BuLi/KOt-Bu/TMP(H) to generate the corresponding dianions 7a–e, which upon oxidative coupling gave the corresponding metacyclophanes 8a–e. This provided unsubstituted cyclophane 8a in 40% yield and 5,13-disubstituted derivatives 8b–d, containing CH3, OCH3 and N(CH3)2 substituents respectively, in comparable yields (Table 2, entries 1–4). In addition, the mono-methoxy substituted derivative 6e was effectively ring closed under our reaction conditions to yield 8e in a 42% yield. In each case, the majority of the remaining material was oligomeric in nature, although it was not characterised. Substrate 6f offered the potential to generate the planar chiral 4-carboxylic acid substituted metacyclophane 8f. This was readily achieved in a 39% yield, with the characteristic NMR aromatic proton signals for C(H)-8/16 observed at 4.21 and 4.18 ppm. The efficient two step synthesis of a C(4)-substituted planar chiral 8f was achieved from inexpensive substrates, under identical reagent conditions for both steps. This compares favourably to the previously reported elaborate seven step synthesis, which was required due to the difficulties of incorporating substituents at the C(4) position after metacyclophane synthesis [27,28]. The resolution of 8f by salt formation with (+)-1-phenylethylamine [27] has previously been accomplished.
The stepwise anti conformation of the metacyclophane 8c was confirmed by single crystal X-ray analysis. Cyclophane 8c crystallised by the slow room temperature evaporation of a diethyl ether solution, into the monoclinic space group P21/n as shown in Figure 4. 8c contains an inversion centre with the co-planar aromatic rings bent into shallow boat forms with an angle of 8.9(1)° from planarity. The intra-annular distance as measured from C(8) to C(16) is as expected for [2.2]metacyclophanes at 2.66(1) Å [29].
![[1860-5397-7-145-4]](/bjoc/content/figures/1860-5397-7-145-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-Ray structure of 8c with thermal ellipsoids drawn at 50% probability level.
Figure 4: X-Ray structure of 8c with thermal ellipsoids drawn at 50% probability level.
Conclusion
A new two-step general approach to [2.2]metacyclophane synthesis was described from substituted m-xylenes. Our strategy employs a selective benzylic metalation and oxidative C–C bond formation for both synthetic operations under LiNK metalation conditions. The synthetic ease of this approach compares favourably with previously reported methods and allows for ready access to potentially useful planar chiral derivatives. Expansion of this strategy to other chiral cyclophane architectures is ongoing and will be reported in due course. Additional synthetic applications of LiNK metalation conditions are also under development.
Supporting Information
| Supporting Information File 1: All experimental details, 1H and 13C NMR spectra for compounds 6a–f and 8a–f and X-ray crystallographic data for 8c. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Whisler, M. C.; MacNeil, S.; Snieckus, V.; Beak, P. Angew. Chem., Int. Ed. 2004, 43, 2206–2225. doi:10.1002/anie.200300590
Return to citation in text: [1] -
Schlosser, M. Angew. Chem., Int. Ed. 2005, 44, 376–393. doi:10.1002/anie.200300645
Return to citation in text: [1] -
Mulvey, R. E. Acc. Chem. Res. 2009, 42, 743–755. doi:10.1021/ar800254y
Return to citation in text: [1] -
Clark, R. D.; Jahangir, A. Org. React. 1995, 47, 1–314. doi:10.1002/0471264180.or047.01
Return to citation in text: [1] -
Clayden, J. Regioselective Synthesis of Organolithiums by Deprotonation. In Organolithiums: Selectivity for Synthesis; Baldwin, J. E.; Williams, R. M., Eds.; Pergamon Press: Oxford, U.K., 2002; pp 9–110.
Return to citation in text: [1] -
Tricotet, T.; Fleming, P.; Cotter, J.; Hogan, A.-M. L.; Strohmann, C.; Gessner, V. H.; O’Shea, D. F. J. Am. Chem. Soc. 2009, 131, 3142–3143. doi:10.1021/ja809941n
Return to citation in text: [1] -
Fleming, P.; O’Shea, D. F. J. Am. Chem. Soc. 2011, 133, 1698–1701. doi:10.1021/ja110234v
Return to citation in text: [1] -
Cram, D. J.; Cram, J. M. Acc. Chem. Res. 1971, 4, 204–213. doi:10.1021/ar50042a003
Return to citation in text: [1] -
Liu, X.; Ma, Y.; Duan, W.; He, F.; Zhao, L.; Song, C. J. Org. Chem. 2011, 76, 1953–1956. doi:10.1021/jo102538y
Return to citation in text: [1] -
Aly, A. A.; Brown, A. B. Tetrahedron 2009, 65, 8055–8089. doi:10.1016/j.tet.2009.06.034
Return to citation in text: [1] -
Vorontsova, N. V.; Rozenberg, V. I.; Sergeeva, E. V.; Vorontsov, E. V.; Starikova, Z. A.; Lyssenko, K. A.; Hopf, H. Chem.–Eur. J. 2008, 14, 4600–4617. doi:10.1002/chem.200701683
Return to citation in text: [1] -
Whelligan, D. K.; Bolm, C. J. Org. Chem. 2006, 71, 4609–4618. doi:10.1021/jo060668h
Return to citation in text: [1] -
Gibson, S. E.; Knight, J. D. Org. Biomol. Chem. 2003, 1, 1256–1269. doi:10.1039/b300717k
Return to citation in text: [1] -
Glotzmann, C.; Langer, E.; Lehner, H.; Schlögl, K. Monatsh. Chem. 1975, 106, 763–777. doi:10.1007/BF00902182
Return to citation in text: [1] -
Glotzmann, C.; Langer, E.; Lehner, H.; Schlögl, K. Tetrahedron Lett. 1975, 9, 675–678. doi:10.1016/S0040-4039(00)71952-6
Return to citation in text: [1] [2] -
Schlögl, K. Top. Curr. Chem. 1984, 125, 27–62.
Return to citation in text: [1] -
Allinger, N. L.; Da Rooge, M. A.; Hermann, R. B. J. Am. Chem. Soc. 1961, 83, 1974–1978. doi:10.1021/ja01469a046
Return to citation in text: [1] -
Akabori, S.; Sato, T.; Hata, K. J. Org. Chem. 1968, 33, 3277–3283. doi:10.1021/jo01272a051
Return to citation in text: [1] -
Higuchi, H.; Tani, K.; Otsubo, T.; Sakata, Y.; Misumi, S. Bull. Chem. Soc. Jpn. 1987, 60, 4027–4036. doi:10.1246/bcsj.60.4027
Return to citation in text: [1] -
Bodwell, G. J.; Houghton, T. J.; Kennedy, J. W. J.; Mannion, M. R. Angew. Chem., Int. Ed. Engl. 1996, 35, 2121–2123. doi:10.1002/anie.199621211
Return to citation in text: [1] -
Vögtle, F.; Neumann, P. Synthesis 1973, 85–103. doi:10.1055/s-1973-22137
Return to citation in text: [1] -
Bates, R. B.; Ogle, C. A. J. Org. Chem. 1982, 47, 3949–3952. doi:10.1021/jo00141a027
Return to citation in text: [1] -
MacNeil, S. L.; Familoni, O. B.; Snieckus, V. J. Org. Chem. 2001, 66, 3662–3670. doi:10.1021/jo001402s
Return to citation in text: [1] -
Nagano, T.; Hayashi, T. Chem. Lett. 2005, 34, 1152–1153.
For the oxidative homo-coupling of Grignard reagents.
Return to citation in text: [1] -
Aluri, B. R. Synlett 2008, 1579–1580. doi:10.1055/s-2008-1072785
Return to citation in text: [1] -
Creger, P. L. J. Am. Chem. Soc. 1970, 92, 1396–1397. doi:10.1021/ja00708a048
Return to citation in text: [1] -
Kainradl, B.; Langer, E.; Lehner, H.; Schlögl, K. Liebigs Ann. Chem. 1973, 766, 16–31. doi:10.1002/jlac.19727660104
Return to citation in text: [1] [2] -
Eberhardt, R.; Glotzmann, C.; Lehner, H.; Schlögl, K. Tetrahedron Lett. 1974, 15, 4365–4638. doi:10.1016/S0040-4039(01)92165-3
Return to citation in text: [1] -
Crystal structure data deposited at the Cambridge Crystallographic Data Centre with deposit number CCDC 825095.
Return to citation in text: [1]
| 1. | Whisler, M. C.; MacNeil, S.; Snieckus, V.; Beak, P. Angew. Chem., Int. Ed. 2004, 43, 2206–2225. doi:10.1002/anie.200300590 |
| 2. | Schlosser, M. Angew. Chem., Int. Ed. 2005, 44, 376–393. doi:10.1002/anie.200300645 |
| 3. | Mulvey, R. E. Acc. Chem. Res. 2009, 42, 743–755. doi:10.1021/ar800254y |
| 4. | Clark, R. D.; Jahangir, A. Org. React. 1995, 47, 1–314. doi:10.1002/0471264180.or047.01 |
| 5. | Clayden, J. Regioselective Synthesis of Organolithiums by Deprotonation. In Organolithiums: Selectivity for Synthesis; Baldwin, J. E.; Williams, R. M., Eds.; Pergamon Press: Oxford, U.K., 2002; pp 9–110. |
| 14. | Glotzmann, C.; Langer, E.; Lehner, H.; Schlögl, K. Monatsh. Chem. 1975, 106, 763–777. doi:10.1007/BF00902182 |
| 15. | Glotzmann, C.; Langer, E.; Lehner, H.; Schlögl, K. Tetrahedron Lett. 1975, 9, 675–678. doi:10.1016/S0040-4039(00)71952-6 |
| 16. | Schlögl, K. Top. Curr. Chem. 1984, 125, 27–62. |
| 9. | Liu, X.; Ma, Y.; Duan, W.; He, F.; Zhao, L.; Song, C. J. Org. Chem. 2011, 76, 1953–1956. doi:10.1021/jo102538y |
| 10. | Aly, A. A.; Brown, A. B. Tetrahedron 2009, 65, 8055–8089. doi:10.1016/j.tet.2009.06.034 |
| 11. | Vorontsova, N. V.; Rozenberg, V. I.; Sergeeva, E. V.; Vorontsov, E. V.; Starikova, Z. A.; Lyssenko, K. A.; Hopf, H. Chem.–Eur. J. 2008, 14, 4600–4617. doi:10.1002/chem.200701683 |
| 12. | Whelligan, D. K.; Bolm, C. J. Org. Chem. 2006, 71, 4609–4618. doi:10.1021/jo060668h |
| 13. | Gibson, S. E.; Knight, J. D. Org. Biomol. Chem. 2003, 1, 1256–1269. doi:10.1039/b300717k |
| 8. | Cram, D. J.; Cram, J. M. Acc. Chem. Res. 1971, 4, 204–213. doi:10.1021/ar50042a003 |
| 29. | Crystal structure data deposited at the Cambridge Crystallographic Data Centre with deposit number CCDC 825095. |
| 6. | Tricotet, T.; Fleming, P.; Cotter, J.; Hogan, A.-M. L.; Strohmann, C.; Gessner, V. H.; O’Shea, D. F. J. Am. Chem. Soc. 2009, 131, 3142–3143. doi:10.1021/ja809941n |
| 7. | Fleming, P.; O’Shea, D. F. J. Am. Chem. Soc. 2011, 133, 1698–1701. doi:10.1021/ja110234v |
| 27. | Kainradl, B.; Langer, E.; Lehner, H.; Schlögl, K. Liebigs Ann. Chem. 1973, 766, 16–31. doi:10.1002/jlac.19727660104 |
| 28. | Eberhardt, R.; Glotzmann, C.; Lehner, H.; Schlögl, K. Tetrahedron Lett. 1974, 15, 4365–4638. doi:10.1016/S0040-4039(01)92165-3 |
| 15. | Glotzmann, C.; Langer, E.; Lehner, H.; Schlögl, K. Tetrahedron Lett. 1975, 9, 675–678. doi:10.1016/S0040-4039(00)71952-6 |
| 27. | Kainradl, B.; Langer, E.; Lehner, H.; Schlögl, K. Liebigs Ann. Chem. 1973, 766, 16–31. doi:10.1002/jlac.19727660104 |
| 22. | Bates, R. B.; Ogle, C. A. J. Org. Chem. 1982, 47, 3949–3952. doi:10.1021/jo00141a027 |
| 23. | MacNeil, S. L.; Familoni, O. B.; Snieckus, V. J. Org. Chem. 2001, 66, 3662–3670. doi:10.1021/jo001402s |
| 24. |
Nagano, T.; Hayashi, T. Chem. Lett. 2005, 34, 1152–1153.
For the oxidative homo-coupling of Grignard reagents. |
| 17. | Allinger, N. L.; Da Rooge, M. A.; Hermann, R. B. J. Am. Chem. Soc. 1961, 83, 1974–1978. doi:10.1021/ja01469a046 |
| 18. | Akabori, S.; Sato, T.; Hata, K. J. Org. Chem. 1968, 33, 3277–3283. doi:10.1021/jo01272a051 |
| 19. | Higuchi, H.; Tani, K.; Otsubo, T.; Sakata, Y.; Misumi, S. Bull. Chem. Soc. Jpn. 1987, 60, 4027–4036. doi:10.1246/bcsj.60.4027 |
| 20. | Bodwell, G. J.; Houghton, T. J.; Kennedy, J. W. J.; Mannion, M. R. Angew. Chem., Int. Ed. Engl. 1996, 35, 2121–2123. doi:10.1002/anie.199621211 |
| 21. | Vögtle, F.; Neumann, P. Synthesis 1973, 85–103. doi:10.1055/s-1973-22137 |
© 2011 Blangetti et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)