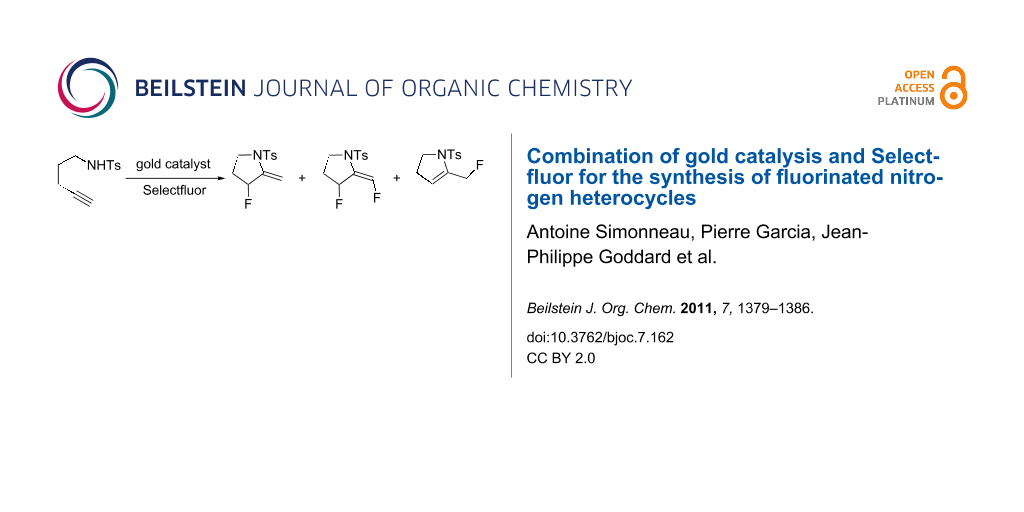
Abstract
We herein report the synthesis of 3-fluoro-2-methylene-pyrrolidine (3a) and -piperidine (3b) from 1,5- and 1,6-aminoalkynes, respectively, using a combination of a gold-catalyzed hydroamination reaction followed by electrophilic trapping of an intermediate cyclic enamine by Selectfluor. Careful attention was paid to the elucidation of the mechanism and Selectfluor was suggested to play the double role of promoting the oxidation of gold(I) to a gold(III) active species and also the electrophilic fluorination of the enamine intermediates.

Graphical Abstract
Introduction
The useful properties of fluorinated compounds in medicinal chemistry have motivated an intense effort towards the synthesis of new molecules bearing fluorine substituents [1,2]. Therefore, the development of a rapid access to C–F bonds is of great importance. Quite recently, in their study on the synthesis α-fluoro ketones, Nevado et al. observed the formation of fluorinated pyrrolidinol obtained by a gold-catalyzed cyclization of a 1,5-aminoalkyne in the presence of Selectfluor (Scheme 1) [3]. These authors proposed that the formation of the C(sp3)–F bond could be explained either by direct fluorination of the enamine resulting from the gold-promoted alkyne hydroamination or by oxidation of the intermediate vinyl gold(I) complex by Selectfluor into a gold(III) fluoride species followed by a reductive elimination.
![[1860-5397-7-162-i1]](/bjoc/content/inline/1860-5397-7-162-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Amino-hydroxyfluorination of alkynes reported by Nevado et al. [2].
Scheme 1: Amino-hydroxyfluorination of alkynes reported by Nevado et al. [2].
Moreover, the formation of C(sp2)–F bonds, either by hydrofluorination of alkynes catalyzed by N-heterocyclic carbene gold(I) complexes [4], or by fluorodeauration of transient vinyl gold species [5,6], has been previously reported in the literature.
On the basis of our recent results on the gold-catalyzed cyclization of enynes [7-10] and allenylhydrazones [11], as well as the studies from Nevado [3], Hammond and Xu [12], Liu and Xu [13], and Liu [14], we were attracted by the possibility to access fluorinated nitrogen heterocycles 2a and 2b by performing subsequently an intramolecular nucleophilic attack of nitrogen onto the gold-activated triple bond on compounds 1a,b in a 5- and 6-exo-dig manner, respectively, and reductive elimination occurring at a vinyl gold(III) fluoride species or bimolecular fluorodeauration (Scheme 2). We also anticipated from this study to gain more insight into the reactivity of gold catalysts/Selectfluor combinations [15-17].
![[1860-5397-7-162-i2]](/bjoc/content/inline/1860-5397-7-162-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed access to fluoromethylene pyrrolidines and piperidines.
Scheme 2: Proposed access to fluoromethylene pyrrolidines and piperidines.
Indeed, pyrrolidine and piperidine skeletons are very attractive ring systems because of their occurrence in numerous biologically active substances, and the design of methodologies allowing the easy introduction of a fluorine atom onto these skeletons could be attractive for medicinal chemists. Recently also, the literature has featured valuable access routes to pyrrolidine promoted by catalytic systems [18-26].
Results and Discussion
Studies on the scope and limitation of the cyclization–fluorination sequence were carried out with Selectfluor as a source of electrophilic fluorine. Readily available 4-methyl-N-(pent-4-ynyl)benzenesulfonamide (1a) was used as a model substrate, and all the reactions were performed in anhydrous acetonitrile as the solvent. Various gold catalysts were screened. The results of the optimization of the cyclization reaction conditions are summarized in Table 1.
Table 1: Gold catalyst influence on the cyclization of 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-162-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | 3a (yield %) | 4a (yield %) | 5a (yield %) |
|---|---|---|---|---|
| 1b | Ph3PAuCl | 75 | 17 | 0 |
| 2c | AuCl | 46 | 0 | 7 |
| 3b | AuCl3 | 14 | 2 | 2 |
| 4 | IPrAuCl | 65 | 13 | 0 |
| 5d | (biphenyl)(t-Bu)2PAuCl | 0 | 0 | 0 |
| 6 | (PhO)3PAuCl | 35 | 7 | 0 |
| 7 | (2,4-di-t-BuPhO)3PAuCl | 35 | 5 | 0 |
| 8 | (t-Bu)3PAuCl | 35 | 6 | 0 |
| 9 | dppm(AuCl)2 | 43 | 0 | 14 |
| 10 | Ph3PAuNTf2 | 35 | 0 | 11 |
| 11 | [Ph3PAu]SbF6 | - | - | - |
aProducts 3a, 4a and 5a were characterized by 1H, 13C and 19F NMR. Compound 4a was obtained as a single (E)-isomer and the configuration of the double bond was confirmed by NOE measurements. bWhen the reaction was performed without Selectfluor, the starting material was recovered unchanged. cReaction time: 5 h.dAn inextricable mixture was obtained.
The reaction of 1a in the presence of commercially available Ph3PAuCl (5 mol %) and Selectfluor (1.1 equiv) in acetonitrile at rt afforded an inseparable mixture of pyrrolidines 3a in 75% yield and 4a in 17% yield (Table 1, entry 1). Difluoro derivative 4a was isolated as a single diastereomer, and its relative stereochemistry was determined by F–F NOE measurement. The expected fluoromethylene tosylpyrrolidine 2a was not detected. When a AuCl complex was used, product 3a was obtained in 46% yield with a new monofluorinated 2-pyrroline 5a in 7% yield (Table 1, entry 2). In the presence of AuCl3, a lower yield of 3a was observed (14%) with trace amounts of 4a and 5a (2% yield each, Table 1, entry 3). The use of the N,N-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) gold(I) chloride as catalyst led to the formation of 3a in 65% yield, together with 13% yield of 4a. Under these conditions, the formation of 5a was not detected (Table 1, entry 4). Gold(I) phosphite catalysts gave 3a and 4a in low yields (Table 1, entries 6 and 7). A similar result was observed with tri(tert-butyl)phosphine gold(I) chloride (Table 1, entry 8). The dinuclear complex, dppm(AuCl)2, led to 3a and 5a in a manner comparable to AuCl (Table 1, entry 9). When we used cationic gold(I) catalyst, Ph3PAuNTf2, 3a was obtained in 35% yield with 11% of 5a (Table 1, entry 10). Finally in the presence of [Ph3PAu]SbF6 a complex mixture of compounds was obtained (Table 1, entry 11). To the best of our knowledge, these fluorinated pyrrolidines 3a, 4a and 2-pyrroline 5a are unknown and could be interesting building blocks for organic synthesis.
Following the previous catalyst screening, we stuck with the use of PPh3AuCl as the catalyst and next investigated the effect of the concentration of 1a and the stoichiometry of Selectfluor (Table 2).
Table 2: Effects of reaction conditions on the Au(I)-catalyzed cyclization of 1a in the presence of Selectfluor.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-162-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | n (equiv) | Substrate concentration (mM) | Temperature | 3a (yield %) | 4a (yield %) |
|---|---|---|---|---|---|
| 1a | 1.5 | 75 | reflux | 25 | 25 |
| 2 | 1.5 | 75 | rt | 47 | 17 |
| 3 | 1.1 | 75 | rt | 54 | 13 |
| 4b | 1.1 | 75 | rt | 0 | 0 |
| 5 | 1.1 | 75 | 5 °C | 17 | 2 |
| 6 | 1.1 | 25 | rt | 75 | 17 |
| 7 | 1.1 | 15 | rt | 73 | 0 |
aReaction time: 1 h. b2 equiv of K2CO3 were added.
Reaction of a 75 mM solution of 1a with Ph3PAuCl (5 mol %) and Selectfluor (1.5 equiv) in acetonitrile under reflux (Table 2, entry 1) afforded an inseparable mixture of 3a and 4a both in 25% yield. When the reaction was performed at room temperature, 3a could be isolated in 47% yield with 17% of 4a (Table 2, entry 2). Using lower amounts of Selectfluor raised the yield of 3a to 54% and 4a to 13% (Table 2, entry 3). In the presence of two equivalents of potassium carbonate the cyclization reaction did not occur (Table 2, entry 4). Lowering the temperature to 5 °C led to a dramatic decrease of the yield of 3a and 4a (Table 2, entry 5). Interestingly, the yield of 3a increased up to 75% when a lower substrate concentration was used (25 mM, Table 2, entry 6). Going to an even more dilute medium resulted in the sole formation of 3a (Table 2, entry 7). It is noteworthy that in the absence of either the gold catalyst or Selectfluor, the starting material was recovered.
The homologue of 1a, compound 1b, was treated with Ph3PAuCl (5 mol %) and Selectfluor (1.1 equiv) in acetonitrile at rt. As expected, the 6-exo-dig cyclization occurred and only led to one compound, 3b, which was isolated in 43% yield (Scheme 3).
![[1860-5397-7-162-i3]](/bjoc/content/inline/1860-5397-7-162-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclization of 1b under standard conditions.
Scheme 3: Cyclization of 1b under standard conditions.
Our mechanistic proposal for the formation of fluorinated pyrrolidines is outlined in Scheme 4. Oxidation of the Au(I) complex by Selectfluor should give the active cationic Au(III) species A. Formation of A is consistent with 19F NMR experiments analogous to those previously described in the literature [12,27]. Thus, upon addition of Selectfluor to PPh3AuCl, a new peak at −181.6 ppm in CD3CN was observed that is characteristic of Au(III) species A [28]. Coordination of 1a to A would lead to complex B in which the coordinated triple bond is activated towards a nucleophilic attack by the NH moiety. The resulting σ-vinyl Au(III) intermediate C could undergo a reductive elimination of its σ-vinyl and F ligands to give 2a, or a protodeauration leading to pyrrolidine 6, which would also rapidly isomerize into 7. Both 6 and 7 under the given reaction conditions would evolve to 3a and 5a.
![[1860-5397-7-162-i4]](/bjoc/content/inline/1860-5397-7-162-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The following experiments were performed to probe the mechanism proposed in Scheme 4. The reaction of a mixture of 6 and 7, in a 2:3 ratio, with PPh3AuCl (5 mol %) and Selectfluor (1 equiv) in acetonitrile at rt during 3 h, led to a mixture of 3a and 5a with yields of 74% and 4%, respectively. In the absence of a gold catalyst, a similar result was obtained (Scheme 5) confirming that Selectfluor itself can react with 6 and 7 to give 3a and 5a, which is consistent with Shreeve’s study on the fluorination of enamines [29].
However, the formation of 5a may also be ascribed to the reductive elimination of C, which leads to 2a, and then further isomerization of the double bond of 2a (Scheme 4). As far as the formation of 4a is concerned, we found that treatment of 3a with one equivalent of Selectfluor in the presence of Ph3PAuCl (5 mol %), led to 4a in 81% yield. Nevertheless, in the absence of Au(I) catalyst, 4a was also formed but with a lower yield of 38% (Scheme 5). These results suggest that the formation of 4a may not exclusively result from the Selectfluor-mediated fluorination of 3a. These findings are consistent with Gouverneur's study [5], which showed a competition between fluorodeauration and protodeauration. In our case, protodeauration appears to be the major, if not the exclusive, pathway.
![[1860-5397-7-162-i5]](/bjoc/content/inline/1860-5397-7-162-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
In a final experiment, 1a was treated with PPh3AuNTf2, without Selectfluor, and dimer 8 was formed in 63% yield along with 6 and 7 in low yields of 2% and 8%, respectively. The formation of 8 could be explained as outlined in Scheme 6. The cationic Au(I) catalyst would promote the cyclization of 1a to the mixture of enamines 6 and 7 as previously observed with cationic Au(III) species (Scheme 4). Activation of the electron rich double bond of 6 or 7 by the cationic Au(I) complex could finally trigger the dimerization and so the formation of 8.
![[1860-5397-7-162-i6]](/bjoc/content/inline/1860-5397-7-162-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Cationic Au(I)-catalyzed reaction of 1a without Selectfluor.
Scheme 6: Cationic Au(I)-catalyzed reaction of 1a without Selectfluor.
Conclusion
In conclusion, we have reported a gold-catalyzed synthesis of fluorinated pyrrolidines from 1,ω-aminoalkynes using Selectfluor as the source of fluorine. This method allows a rapid, efficient and mild conversion of readily available aminoalkynes into valuable nitrogen heterocycles substituted by a fluorine atom in position 3 of the ring. This could certainly be applied to the synthesis of biologically relevant substrates. Current efforts are being made in this direction and a more exemplified study will be reported in due course.
Experimental
General methods
Acetonitrile was distilled over calcium hydride. Other reagents were commercially available and used without further purification. Thin layer chromatography (TLC) was performed on Merck 60 F254 silica gel. Acros aluminium oxide, basic, Brockmann I, 50–200 µm, 60A was used for column chromatography. NMR spectra (1H, 13C, DEPT, COSY, HMQC, HMBC, NOE) were recorded at room temperature at 300 or 400 MHz on a Bruker AVANCE spectrometer. Chemical shifts are given in ppm, referenced to the residual proton resonance of the solvents (δ = 7.26 for CHCl3) or to the residual carbon resonance of the solvent (δ = 77.16 for CDCl3). Coupling constants (J) are given in Hertz (Hz). The terms m, s, d, t and q refer to multiplet, singlet, doublet, triplet and quartet; br means that the signal is broad. When possible, 1H and 13C signals were assigned on the basis of DEPT and 2D NMR (COSY, HMBC) experiments. Low-resolution mass spectra (MS) and high-resolution mass spectra (HRMS) were measured on a Bruker MicroTOF mass spectrometer. Infrared spectra (IR) were recorded on a Bruker Tensor 27 spectrometer and melting points were measured on a Wagner & Munz HEIZBANK Kofler bench.
General procedure for the synthesis of precursors
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-162-i9.svg?max-width=637&scale=1.18182)
To a cold solution (0 °C) of the starting alcohol in MeCN, MsCl and Et3N were successively added, and the mixture was warmed to rt. After 1 h, K2CO3 and TsNH2 were added and the mixture was warmed at 80 °C overnight. Once back to rt, the mixture was directly purified by flash chromatography being eluted first with petroleum ether and then with petroleum ether/ethyl acetate 9:1.
Spectral data of cyclization precursors
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-162-i10.svg?max-width=637&scale=1.18182)
1a. In agreement with the literature data [30,31].
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-162-i11.svg?max-width=637&scale=1.18182)
1b. In agreement with the literature data [31,32].
General procedure for the cyclization reaction
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-162-i12.svg?max-width=637&scale=1.18182)
Into an oven-dried Schlenk apparatus, the Selectfluor (0.33 mmol, 117 mg, 1.1 equiv) and the gold catalyst (16 µmol, 0.05 equiv) were loaded under a flow of argon. These solids were dried under vacuum at 70–80 °C for 2 h. The aminoalkyne (0.30 mmol, 1 equiv) was then added, followed by anhydrous MeCN (12 mL), under a flow of argon. The mixture was stirred at rt until complete consumption of the starting material was observed by TLC. The reaction was then filtered on a short plug of basic alumina. After removal of the solvents under reduced pressure, the crude product was purified by flash column chromatography on alumina with pentane/ethyl acetate 85:15 as eluent.
Spectral data of cyclic products
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-162-i13.svg?max-width=637&scale=1.18182)
3a. White solid, mp 102 °C; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 5.38 (d, J = 5.2 Hz, 1H), 5.09 (ddd, JH-F = 54.6, JH-H = 4.2, 1.9 Hz, 1H), 4.76 (d, J = 6.0 Hz, 1H), 3.88–3.82 (m, 1H), 3.63 (td, J = 9.8, 6.5 Hz, 1H), 2.42 (s, 3H), 2.14–1.87 (m, 2H); 19F NMR (376.5 MHz, CDCl3) δ −168.4 (m, 1F); 13C NMR (101 MHz, CDCl3) δ 144.5 (C), 143.2 (d, JC-F = 15.4 Hz, C), 134.2 (C), 129.7 (2CH), 127.6 (2CH), 97.2 (d, JC-F = 8.4 Hz, CH2), 93.7 (d, JC-F = 178.5 Hz, CH), 48.6 (CH2), 29.5 (d, JC-F = 22.3 Hz, CH2), 21.7 (CH3); IR (neat) 2358, 1652, 1338, 1251, 1156, 1085, 1000, 866, 814, 651 cm−1; HRMS (m/z): [M + Na]+ calcd for C12H14FNO2S, 278.0621; found, 278.0632.
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-162-i14.svg?max-width=637&scale=1.18182)
5a. Isolated as a minor product in mixture with 3a; 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 5.39–5.37 (m, 1H), 5.22 (d, JH-F = 46.8 Hz, 2H), 3.78 (t, J = 8.9 Hz, 2H), 2.43 (s, 3H), 2.33–2.22 (m, 2H); 19F NMR (376 MHz, CDCl3) δ −213.6 to −213.9 (m, 1F); 13C NMR (101 MHz, CDCl3) δ 144.1 (C), 139.5 (d, JC-F = 20.7 Hz, C), 134.1 (C), 130.1 (2CH), 127.8 (2CH), 115.4 (d, JC-F = 8.1 Hz, CH), 78.5 (d, JC-F = 167.4 Hz, CH2), 50.4 (CH2), 27.7 (CH2), 21.7 (CH3).
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-162-i15.svg?max-width=637&scale=1.18182)
4a. White solid, mp 68 °C; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.3 Hz, 2H), 7.39 (dd, JH-F = 80.7, JH-H = 6.1 Hz, 1H), 7.32 (d, J = 8.1 Hz, 2H), 5.63 (dt, JH-F = 53.8 Hz, JH-H = 3.2 Hz, 1H), 3.83 (t, J = 9.2 Hz, 1H), 3.48 (ddd, J = 11.1, 10.1, 6.2 Hz, 1H), 2.43 (s, 3H), 2.09 (ddd, J = 17.5, 14.4, 6.1 Hz, 1H), 1.97–1.76 (m, 1H); 19F NMR (376 MHz, CDCl3) δ −149.5 (dd, JH-F = 80.7, JF-F = 8.9 Hz, 1F), −171.6 to −173.7 (m, 1F); 13C NMR (101 MHz, CDCl3) δ 144.8 (C), 142.0 (dd, JC-F = 250.4, 10.1 Hz, CH), 133.3 (C), 129.9 (2CH), 128.2 (C), 127.8 (2CH), 88.1 (dd, JC-F = 178.7, 3.5 Hz, CHF), 48.7 (CH2), 30.4 (d, JC-F = 23.1 Hz, CH2), 21.8 (CH3); IR (neat) 2359, 1597, 1353, 1163, 1133, 1090, 1060, 1011, 964, 814, 664 cm−1; HRMS (m/z): [M + Na]+ calcd for C12H13F2NO2S, 296.0527; found, 296.0535.
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-162-i16.svg?max-width=637&scale=1.18182)
3b. Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 5.35 (s, 1H), 5.16 (s, 1H), 4.70 (dt, JH-F = 49.6 Hz, JH-H = 5.3 Hz, 1H), 3.68–3.54 (m, 2H), 2.42 (s, 3H), 1.97–1.81 (m, 2H), 1.73 (m, 1H), 1.61–1.48 (m, 1H); 19F NMR (376 MHz, CDCl3) δ −173.4 (m, 1F); 13C NMR (101 MHz, CDCl3) δ 143.7 (C), 140.5 (d, JC-F = 19.0 Hz, C), 137.3 (C), 129.7 (2CH), 127.7 (2CH), 110.7 (d, JC-F = 7.7 Hz, CH2), 88.4 (d, JC-F = 178.6 Hz, CHF), 46.9 (CH2), 30.7 (d, JC-F = 21.7 Hz, CH2), 21.7 (CH3), 20.9 (d, JC-F = 5.4 Hz, CH2); IR (neat) 1647, 1598, 1451, 1340, 1157, 1098, 1057, 950, 908, 814, 690, 653 cm−1; HRMS (m/z): [M + Na]+ calcd for C13H16FNO2S, 292.0778; found, 292.0776.
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-162-i17.svg?max-width=637&scale=1.18182)
8. White solid, mp 66 °C; 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 5.2 Hz, 2H), 7.28 (d, J = 6.3 Hz, 2H), 5.41 (br s, 1H), 3.89–3.69 (m, 2H), 3.54–3.46 (m, 1H), 3.30–3.23 (m, 3H), 2.66–2.52 (m, 1H), 2.44 (s, 3H), 2.42 (s, 3H), 2.01–1.87 (m, 3H), 1.78–1.58 (m, 2H), 1.41 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 143.9 (C), 142.8 (C), 140.9 (C), 138.8 (C), 134.1 (C), 129.7 (2CH), 129.5 (2CH), 127.8 (2CH), 127.3 (2CH), 119.5 (CH), 68.20 (C), 51.3 (CH2), 49.9 (CH2), 39.6 (CH2), 39.2 (CH2), 27.7 (CH2), 26.6 (CH3), 22.7 (CH2), 21.7 (CH3), 21.6 (CH3); IR (neat) 1452, 1332, 1154, 1089, 1000, 811, 655 cm−1; HRMS (m/z): [M + Na]+ calcd for C24H30N2O4S2, 497.1539; found, 497.1517.
Synthesis of cyclic enamines
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-162-i18.svg?max-width=637&scale=1.18182)
Cyclic enamines were synthesized in 59% yield (endo/exo 2:3) according to a literature procedure [32]. Spectral data matched those reported.
General procedure for the fluorination reactions of the enamines
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-162-i19.svg?max-width=637&scale=1.18182)
Conditions A: In an oven-dried Schlenk apparatus the Selectfluor (0.17 mmol, 1 equiv) was loaded under a flow of argon. This was then dried under vacuum at 70–80 °C for 2 h. The mixture of cyclic enamines (0.17 mmol, 40 mg, 1 equiv) was then added, followed by anhydrous MeCN (7 mL), under a flow of argon. The mixture was stirred at rt until complete consumption of the starting material was observed by TLC. The reaction mixture was then filtered on a short plug of basic alumina. After removal of the solvents under reduced pressure, the crude product was purified by flash column chromatography on alumina with pentane/ethyl acetate 85:15 as eluent.
Conditions B: In an oven-dried Schlenk apparatus, the Selectfluor (0.33 mmol, 117 mg, 1.1 equiv) and triphenylphosphine gold chloride (16 µmol, 7.4 mg, 0.05 equiv) were loaded under a flow of argon. These solids were dried under vacuum at 70–80 °C for 2 h. A mixture of cyclic enamines (0.3 mmol, 72 mg, 1 equiv) was then added, followed by anhydrous MeCN (12 mL), under a flow of argon. The mixture was stirred at rt until complete consumption of the starting material was observed by TLC. The reaction was then filtered on a short plug of basic alumina. After removal of the solvents under reduced pressure, the crude product was purified by flash column chromatography on alumina with pentane/ethyl acetate 85:15 as eluent.
Supporting Information
| Supporting Information File 1: 1H, 13C, 19F NMR spectra of products 3a, 4a, 5a, 3b and 8. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004.
Return to citation in text: [1] -
Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, 2009. doi:10.1002/9781444312096
Return to citation in text: [1] [2] -
De Haro, T.; Nevado, C. Adv. Synth. Catal. 2010, 352, 2767–2772. doi:10.1002/adsc.201000559
Return to citation in text: [1] [2] -
Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P. J. Am. Chem. Soc. 2007, 129, 7736–7737. doi:10.1021/ja0723784
Return to citation in text: [1] -
Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2008, 47, 7927–7930. doi:10.1002/anie.200802162
Return to citation in text: [1] [2] -
de Haro, T.; Nevado, C. Chem. Commun. 2011, 47, 248–249. doi:10.1039/C002679D
Return to citation in text: [1] -
Moreau, X.; Goddard, J.-P.; Bernard, M.; Lemière, G.; López-Romero, J. M.; Mainetti, E.; Marion, N.; Mouriès, V.; Thorimbert, S.; Fensterbank, L.; Malacria, M. Adv. Synth. Catal. 2008, 350, 43–48. doi:10.1002/adsc.200700356
Return to citation in text: [1] -
Marion, N.; Lemière, G.; Correa, A.; Costabile, C.; Ramón, R. S.; Moreau, X.; de Frémont, P.; Dahmane, R.; Hours, A.; Lesage, D.; Tabet, J.-C.; Goddard, J.-P.; Gandon, V.; Cavallo, L.; Fensterbank, L.; Malacria, M.; Nolan, S. P. Chem.–Eur. J. 2009, 15, 3243–3260. doi:10.1002/chem.200801387
Return to citation in text: [1] -
Harrak, Y.; Makhlouf, M.; Azzaro, S.; Mainetti, E.; Lopez Romero, J. M.; Cariou, K.; Gandon, V.; Goddard, J.-P.; Malacria, M.; Fensterbank, L. J. Organomet. Chem. 2011, 696, 388–399. doi:10.1016/j.jorganchem.2010.10.016
Return to citation in text: [1] -
Harrak, Y.; Simmonneau, A.; Malacria, M.; Gandon, V.; Fensterbank, L. Chem. Commun. 2010, 46, 865–867. doi:10.1039/B919240A
Return to citation in text: [1] -
Benedetti, E.; Lemière, G.; Chapellet, L.-L.; Penoni, A.; Palmisano, G.; Malacria, M.; Goddard, J.-P.; Fensterbank, L. Org. Lett. 2010, 12, 4396–4399. doi:10.1021/ol101889h
Return to citation in text: [1] -
Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593
Return to citation in text: [1] [2] -
Qian, J.; Liu, Y.; Zhu, J.; Jiang, B.; Xu, Z. Org. Lett. 2011, 13, 4220–4223. doi:10.1021/ol201555z
Return to citation in text: [1] -
Xu, T.; Mu, X.; Peng, H.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 8176–8179. doi:10.1002/anie.201103225
Return to citation in text: [1] -
Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054
Return to citation in text: [1] -
Engle, K. M.; Mei, T.-S.; Wang, X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2011, 50, 1478–1491. doi:10.1002/anie.201005142
Return to citation in text: [1] -
Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem.–Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736
Return to citation in text: [1] -
Ney, J. E.; Wolfe, J. P. Angew. Chem., Int. Ed. 2004, 43, 3605–3608. doi:10.1002/anie.200460060
Return to citation in text: [1] -
Hong, S.; Marks, T. J. Acc. Chem. Res. 2004, 37, 673–686. doi:10.1021/ar040051r
Return to citation in text: [1] -
Brenzovich, W. E.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739
Return to citation in text: [1] -
Hannedouche, J.; Collin, J.; Trifonov, A.; Schulz, E. J. Organomet. Chem. 2011, 696, 255–262. doi:10.1016/j.jorganchem.2010.09.013
Return to citation in text: [1] -
Trost, B. M.; Maulide, N.; Livingston, R. C. J. Am. Chem. Soc. 2008, 130, 16502–16503. doi:10.1021/ja807696e
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536–6544. doi:10.1039/C1CC10780A
Return to citation in text: [1] -
Hirano, K.; Inaba, Y.; Takahashi, N.; Shimano, M.; Oishi, S.; Fujii, N.; Ohno, H. J. Org. Chem. 2011, 76, 1212–1227. doi:10.1021/jo102507c
Return to citation in text: [1] -
Peng, H.; Liu, G. Org. Lett. 2011, 13, 772–775. doi:10.1021/ol103039x
Return to citation in text: [1] -
Yeom, H.-S.; So, E.; Shin, S. Chem.–Eur. J. 2011, 17, 1764–1767. doi:10.1002/chem.201002863
Return to citation in text: [1] -
Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n
Return to citation in text: [1] -
19F NMR monitoring of the reaction of an equimolar mixture of Selectfluor and PPh3AuCl in CD3CN indicated that it is a rather slow process. After 3 h at rt, a 5:1 Selectrfluor/A ratio was obtained; after 5 h, this ratio became 4:1 and after 24 h, gold nanoparticles were observed and signals of A vanished. This precluded the isolation of species A and its further use as a reagent.
Return to citation in text: [1] -
Peng, W.; Shreeve, J. M. J. Org. Chem. 2005, 70, 5760–5763. doi:10.1021/jo0506837
Return to citation in text: [1] -
Schomaker, J. M.; Geiser, A. R.; Huang, R.; Borhan, B. J. Am. Chem. Soc. 2007, 129, 3794–3795. doi:10.1021/ja068077w
Return to citation in text: [1] -
Garcia, P.; Evanno, Y.; George, P.; Sevrin, M.; Ricci, G.; Malacria, M.; Aubert, C.; Gandon, V. Org. Lett. 2011, 13, 2030–2033. doi:10.1021/ol200417p
Return to citation in text: [1] [2] -
Fix, S. R.; Brice, J. L.; Stahl, S. S. Angew. Chem., Int. Ed. 2002, 41, 164–166. doi:10.1002/1521-3773(20020104)41:1<164::AID-ANIE164>3.0.CO;2-B
Return to citation in text: [1] [2]
| 31. | Garcia, P.; Evanno, Y.; George, P.; Sevrin, M.; Ricci, G.; Malacria, M.; Aubert, C.; Gandon, V. Org. Lett. 2011, 13, 2030–2033. doi:10.1021/ol200417p |
| 32. | Fix, S. R.; Brice, J. L.; Stahl, S. S. Angew. Chem., Int. Ed. 2002, 41, 164–166. doi:10.1002/1521-3773(20020104)41:1<164::AID-ANIE164>3.0.CO;2-B |
| 5. | Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2008, 47, 7927–7930. doi:10.1002/anie.200802162 |
| 30. | Schomaker, J. M.; Geiser, A. R.; Huang, R.; Borhan, B. J. Am. Chem. Soc. 2007, 129, 3794–3795. doi:10.1021/ja068077w |
| 31. | Garcia, P.; Evanno, Y.; George, P.; Sevrin, M.; Ricci, G.; Malacria, M.; Aubert, C.; Gandon, V. Org. Lett. 2011, 13, 2030–2033. doi:10.1021/ol200417p |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. |
| 2. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, 2009. doi:10.1002/9781444312096 |
| 5. | Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2008, 47, 7927–7930. doi:10.1002/anie.200802162 |
| 6. | de Haro, T.; Nevado, C. Chem. Commun. 2011, 47, 248–249. doi:10.1039/C002679D |
| 28. | 19F NMR monitoring of the reaction of an equimolar mixture of Selectfluor and PPh3AuCl in CD3CN indicated that it is a rather slow process. After 3 h at rt, a 5:1 Selectrfluor/A ratio was obtained; after 5 h, this ratio became 4:1 and after 24 h, gold nanoparticles were observed and signals of A vanished. This precluded the isolation of species A and its further use as a reagent. |
| 4. | Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P. J. Am. Chem. Soc. 2007, 129, 7736–7737. doi:10.1021/ja0723784 |
| 29. | Peng, W.; Shreeve, J. M. J. Org. Chem. 2005, 70, 5760–5763. doi:10.1021/jo0506837 |
| 2. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, 2009. doi:10.1002/9781444312096 |
| 18. | Ney, J. E.; Wolfe, J. P. Angew. Chem., Int. Ed. 2004, 43, 3605–3608. doi:10.1002/anie.200460060 |
| 19. | Hong, S.; Marks, T. J. Acc. Chem. Res. 2004, 37, 673–686. doi:10.1021/ar040051r |
| 20. | Brenzovich, W. E.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 21. | Hannedouche, J.; Collin, J.; Trifonov, A.; Schulz, E. J. Organomet. Chem. 2011, 696, 255–262. doi:10.1016/j.jorganchem.2010.09.013 |
| 22. | Trost, B. M.; Maulide, N.; Livingston, R. C. J. Am. Chem. Soc. 2008, 130, 16502–16503. doi:10.1021/ja807696e |
| 23. | Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536–6544. doi:10.1039/C1CC10780A |
| 24. | Hirano, K.; Inaba, Y.; Takahashi, N.; Shimano, M.; Oishi, S.; Fujii, N.; Ohno, H. J. Org. Chem. 2011, 76, 1212–1227. doi:10.1021/jo102507c |
| 25. | Peng, H.; Liu, G. Org. Lett. 2011, 13, 772–775. doi:10.1021/ol103039x |
| 26. | Yeom, H.-S.; So, E.; Shin, S. Chem.–Eur. J. 2011, 17, 1764–1767. doi:10.1002/chem.201002863 |
| 3. | De Haro, T.; Nevado, C. Adv. Synth. Catal. 2010, 352, 2767–2772. doi:10.1002/adsc.201000559 |
| 12. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593 |
| 27. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 12. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2010, 49, 7247–7252. doi:10.1002/anie.201003593 |
| 14. | Xu, T.; Mu, X.; Peng, H.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 8176–8179. doi:10.1002/anie.201103225 |
| 3. | De Haro, T.; Nevado, C. Adv. Synth. Catal. 2010, 352, 2767–2772. doi:10.1002/adsc.201000559 |
| 15. | Hashmi, A. S. K.; Ramamurthi, T. D.; Rominger, F. J. Organomet. Chem. 2009, 694, 592–597. doi:10.1016/j.jorganchem.2008.11.054 |
| 16. | Engle, K. M.; Mei, T.-S.; Wang, X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2011, 50, 1478–1491. doi:10.1002/anie.201005142 |
| 17. | Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem.–Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736 |
| 11. | Benedetti, E.; Lemière, G.; Chapellet, L.-L.; Penoni, A.; Palmisano, G.; Malacria, M.; Goddard, J.-P.; Fensterbank, L. Org. Lett. 2010, 12, 4396–4399. doi:10.1021/ol101889h |
| 32. | Fix, S. R.; Brice, J. L.; Stahl, S. S. Angew. Chem., Int. Ed. 2002, 41, 164–166. doi:10.1002/1521-3773(20020104)41:1<164::AID-ANIE164>3.0.CO;2-B |
| 7. | Moreau, X.; Goddard, J.-P.; Bernard, M.; Lemière, G.; López-Romero, J. M.; Mainetti, E.; Marion, N.; Mouriès, V.; Thorimbert, S.; Fensterbank, L.; Malacria, M. Adv. Synth. Catal. 2008, 350, 43–48. doi:10.1002/adsc.200700356 |
| 8. | Marion, N.; Lemière, G.; Correa, A.; Costabile, C.; Ramón, R. S.; Moreau, X.; de Frémont, P.; Dahmane, R.; Hours, A.; Lesage, D.; Tabet, J.-C.; Goddard, J.-P.; Gandon, V.; Cavallo, L.; Fensterbank, L.; Malacria, M.; Nolan, S. P. Chem.–Eur. J. 2009, 15, 3243–3260. doi:10.1002/chem.200801387 |
| 9. | Harrak, Y.; Makhlouf, M.; Azzaro, S.; Mainetti, E.; Lopez Romero, J. M.; Cariou, K.; Gandon, V.; Goddard, J.-P.; Malacria, M.; Fensterbank, L. J. Organomet. Chem. 2011, 696, 388–399. doi:10.1016/j.jorganchem.2010.10.016 |
| 10. | Harrak, Y.; Simmonneau, A.; Malacria, M.; Gandon, V.; Fensterbank, L. Chem. Commun. 2010, 46, 865–867. doi:10.1039/B919240A |
| 13. | Qian, J.; Liu, Y.; Zhu, J.; Jiang, B.; Xu, Z. Org. Lett. 2011, 13, 4220–4223. doi:10.1021/ol201555z |
© 2011 Simonneau et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)