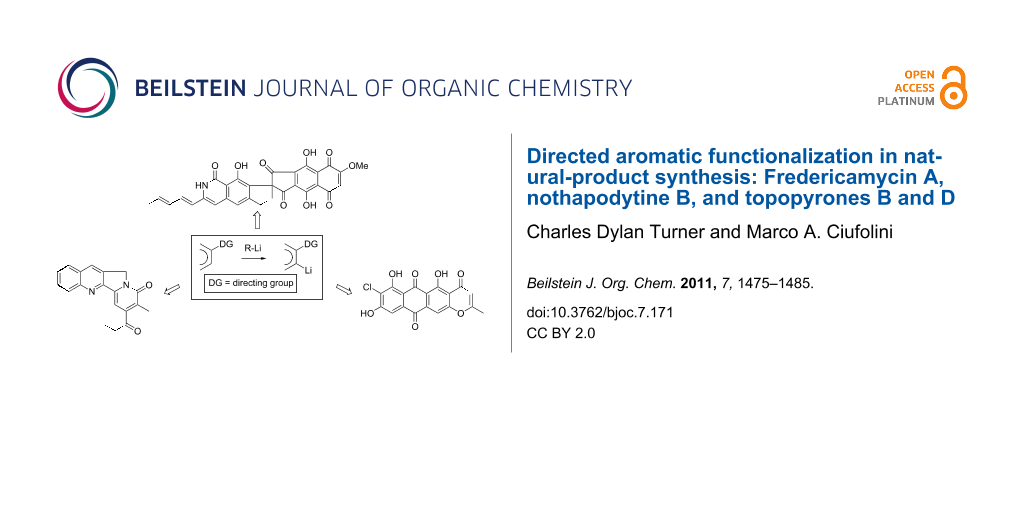
Abstract
This is a review of our efforts toward the synthesis of a group of natural products that display noteworthy biological activity: Fredericamycin A, nothapodytine B, and topopyrones B and D. In each case, directed aromatic functionalization methodology greatly facilitated the assembly of the key molecular subunits.

Graphical Abstract
Review
Our laboratory is primarily interested in the total synthesis of natural products, and does not conduct research on directed aromatic functionalization ("DAF") per se. On numerous occasions, however, DAF technology has been key to the success of specific synthetic endeavors in our group. Herein, we illustrate the application of such techniques to three representative problems that we have addressed over the years: The syntheses of fredericamycin A, nothapodytine B, and topopyrones B and D.
Fredericamycin A
This initial portion of the present review recounts the first project that the senior author of this paper launched as an independent academic in 1984. A structurally novel natural product by the name of fredericamycin A (1, Scheme 1) had been discovered only two years prior and determined to be strongly cytotoxic [1-5]: A finding that spawned a flurry of activity in the synthetic arena. Indeed, seven total syntheses [6-16] and numerous approaches [17-30] have been described since. Fredericamycin seemed to be a superb vehicle to address an issue for which no solution existed at that time: The arylation of stabilized enolates. Indeed, the retrosynthetic analysis illustrated in Scheme 1 suggests that 1 could result from the cyclization of 3, which in turn would ensue through the union of fragments 4 and 5. These two subunits were prepared by DAF technology [31].
![[1860-5397-7-171-i1]](/bjoc/content/inline/1860-5397-7-171-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structure and retrosynthetic analysis of fredericamycin A.
Scheme 1: Structure and retrosynthetic analysis of fredericamycin A.
The starting point for the assembly of an appropriate variant of 4 was diethylamide 6 (Scheme 2), which underwent smooth Beak–Snieckus-type ortho-deprotonation [32-35] with the sec-BuLi–TMEDA complex and consequent borylation in high yield. Oxidation of the ensuing 7 to phenol 8 and O-protection served as a prelude to a second ortho-deprotonation en route to methyl derivative 10. A third round of sec-BuLi–TMEDA treatment induced highly selective deprotonation of the methyl group, and in keeping with the observations of Kelly [6,7], the intervening anion was intercepted by diethoxyacetonitrile [36] to furnish isoquinolone 11 directly. Relative to 4, compound 11 lacks an iodo substituent, which was introduced by reaction of the free aldehyde 12 with I2 and Cu(OAc)2 in refluxing AcOH [37]. Only the unprotected 12 performed satisfactorily in this step, which, unfortunately, afforded an essentially 1:1 mixture of the desired phenolic ortho-iodide 13 and the corresponding para-isomer. These were readily separated after reprotection of the aldehyde and of the phenol, i.e., at the stage of 14. Notice that in the course of the reaction the primary OH group in 12 was converted into an acetate ester (Fischer-type esterification). Compound 15, a form of 4 suitable for the conduction of subsequent operations, was secured by deacetylation of 14 and Swern oxidation.
![[1860-5397-7-171-i2]](/bjoc/content/inline/1860-5397-7-171-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Assembly of the isoquinolone segment of fredericamycin.
Scheme 2: Assembly of the isoquinolone segment of fredericamycin.
Fragment 5 was best produced in the guise of compound 20, the construction of which also relied upon DAF methodology. Thus, ortho-deprotonation of diethylamide 16 (Scheme 3) and carboxylation afforded anhydride 17 directly after workup with 12 N aqueous HCl. Fischer esterification and condensation of the resultant 18 with diethyl succinate, according to Kelly [6,7], afforded 19, which was then elaborated to a 1:1 mixture of regioisomers of naphthalide 20. It will be seen shortly that the formation of regioisomers at this stage is inconsequential.
![[1860-5397-7-171-i3]](/bjoc/content/inline/1860-5397-7-171-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of a naphthalide precursor to the quinoid moiety of fredericamycin.
Scheme 3: Synthesis of a naphthalide precursor to the quinoid moiety of fredericamycin.
Parallel work had concentrated on defining a protocol for the merger of the two fragments, and especially for the conduct of the crucial, and the time highly novel, arylation step. The first issue, the more tractable of the two, was addressed by converting 10 into aldehyde 23 as a model of the more elaborate 15 (Scheme 4). Ironically, and contrary to the case of 12, the iodination of this simpler system proceeded with superb ortho-selectivity to furnish 21 as the sole product. The aldehyde underwent tandem addition of the anion of simple phthalide (a model for 20; prepared from the parent compound by deprotonation with LDA) and dehydration via mesylate 26 to provide 27, which upon reaction with EtOLi (EtOH + BuLi) in THF rearranged to diketone 28. Experiments involving 28, as well as simpler model systems [38], revealed that the few techniques of enolate arylation then known were completely ineffective for the desired transformation. Remarkably, however, heating a DMF solution of the preformed sodium enolate of 28 (NaH) in the presence of Pd(PPh3)4 triggered cyclization to 29 in a very satisfactory 76% yield [39].
![[1860-5397-7-171-i4]](/bjoc/content/inline/1860-5397-7-171-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Palladium-mediated cyclization of a fredericamycin model system.
Scheme 4: Palladium-mediated cyclization of a fredericamycin model system.
On the basis of these results, the anion of naphthalide 20 was condensed with aldehyde 15 following the same protocol to yield compound 32 (Scheme 5). A distressing observation was made at this juncture: The base-promoted transposition of 32 to 33 was complicated by the unexpected propensity of the diketone to react with atmospheric oxygen to form the corresponding 2-hydroxy derivative 34. This transformation was virtually instantaneous when the anion of 33 was exposed to the atmosphere, whereas 33 itself was completely hydroxylated in about 30 minutes at room temperature. Silica gel appeared to act as an effective catalyst for the process, further complicating purification, which ultimately had to be carried out in a glove box.
![[1860-5397-7-171-i5]](/bjoc/content/inline/1860-5397-7-171-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the precursor of fredericamycin and the facile air oxidation thereof.
Scheme 5: Synthesis of the precursor of fredericamycin and the facile air oxidation thereof.
Not without some difficulty, a batch of material consisting largely of 33 was subjected to the palladium-mediated cyclization reaction. This led to the formation of compound 35 in about 60% yield (based on 1H NMR; Scheme 6). A late intermediate in the Kelly synthesis of 1 [6,7] is structurally very similar to 35. Consequently, the preparation of 35 corresponds to a formal synthesis of fredericamycin A [31]. In the years since, Pd-mediated arylation reactions of enolates have been extensively developed and improved, especially by Buchwald [40-43] and Hartwig [44-46].
![[1860-5397-7-171-i6]](/bjoc/content/inline/1860-5397-7-171-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formal synthesis of fredericamycin A.
Scheme 6: Formal synthesis of fredericamycin A.
Nothapodytine
Our group cultivates a long-standing interest in furan chemistry, especially in connection with a process that we describe as the “aza-Achmatowicz reaction” [47-52]: A transformation that has attracted the attention of a number of researchers worldwide [53-62]. In the course of such endeavors, we have often resorted to the facile C-2 deprotonation of furan as a means to generate appropriate derivatives. This is of course a special case of directed aromatic functionalization. A significant example of this chemistry was key to our synthesis of nothapodytine B, 36 (Figure 1) [63]. This substance is also known as mappicine ketone, in that it is an oxidized form of the alkaloid, mappicine [64], and indeed it can be converted into the latter by NaBH4 reduction of the ethyl ketone. A brief historical aside: The term “mappicine ketone” was apparently introduced by Kametani [65], who synthesized 36 in the course of investigations directed toward mappicine, and this more than 20 years before 36 was itself determined to be a natural product. In any event, nothapodytine exhibits interesting antiviral properties [66,67] and it has been the target of a number of total syntheses [68-74] and synthetic studies [75-79].
![[1860-5397-7-171-1]](/bjoc/content/figures/1860-5397-7-171-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The central problem we wished to address was the preparation of the 3-alkyl-2-pyridone moiety of 36 by an unusual [3 + 3] construction developed in our laboratory [80]. This chemistry (Scheme 7) promotes the condensation of a cyanoacetamide 37, with an enone or enal 38, in the presence of t-BuOK in DMSO. Under anoxic conditions, a series of events, culminating in the elimination of HCN from a presumed dianion intermediate, leads to the formation of pyridones 39a, wherein R3 may be H, alkyl, or aryl. Conduct of the reaction with plain cyanoacetamide (cf. 37, R3 = H) under an oxygen atmosphere produces cyanopyridones 39b instead.
![[1860-5397-7-171-i7]](/bjoc/content/inline/1860-5397-7-171-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
In keeping with a principle introduced during our work on camptothecin [81-83], the five-membered ring of 36 was imagined to result upon acid treatment of 40 (Scheme 8) [84], which in turn could be assembled by using the chemistry of Scheme 8 on substrate 41, provided that the more electrophilic quinolyl ketone would direct an initial conjugate addition of the anion of a suitable cyanoacetamide to its own β-position. A particularly direct way to produce 41 seemed to be the oxidative cleavage of the furan ring in 42, which thus became the first subgoal of our study. In that regard, we elected to prepare 42 by Suzuki coupling [85] between a furylboronic acid and a 2-chloroquinoline [86].
![[1860-5397-7-171-i8]](/bjoc/content/inline/1860-5397-7-171-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Retrosynthetic logic for nothapodytine B.
Scheme 8: Retrosynthetic logic for nothapodytine B.
The (relative) acidity of the C-2 position of furan is such that n-BuLi without added TMEDA suffices to induce lithiation. Accordingly, treatment of commercial 2-ethylfuran with n-BuLi in THF at 0 °C and cannulation of the resulting mixture into a solution of trimethyl borate in THF afforded boronic acid 44 (Scheme 9) in 90% yield after the customary aqueous workup [87]. This material underwent smooth Suzuki coupling with the known 45 [81-83], and the action of aqueous NBS [88] upon the resultant 42 delivered the requisite 41 in 87% yield.
![[1860-5397-7-171-i9]](/bjoc/content/inline/1860-5397-7-171-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Preparation of a key nothapodytine fragment.
Scheme 9: Preparation of a key nothapodytine fragment.
Happily, it transpired that the conjugate addition chemistry of 41 is indeed controlled exclusively by the quinolyl ketone. This enabled the conduct of our pyridone-forming sequence, which in the present case, however, had to be implemented in the slightly modified form seen in Scheme 10. Thus, Michael addition of 2-methyl cyanoacetamide converted 41 into a mixture of diastereomers of hemiamidals 47 and 48. Attempts to force this mixture to advance to the desired pyridone under basic conditions yielded uniformly unsatisfactory results. This was due in part to undesired base-promoted reactions of the enolizable ketone segments, but also to the slow rate of interconversion of 47 and 48 (only the former can produce the desired pyridone). Experiment revealed that it was best to carry out the Michael step with DBU in pyridine, followed by addition of Ac2O and warming to 80 °C for an extended period of time. Under these conditions, a mixture of 2-acetoxypyridines 49 and 50 was obtained. Both compounds were transformed into 36 [89] upon exposure to HBr in CF3CH2OH (Boger conditions [68,69]).
![[1860-5397-7-171-i10]](/bjoc/content/inline/1860-5397-7-171-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Total synthesis of nothapodytine B.
Scheme 10: Total synthesis of nothapodytine B.
Topopyrones B and D
Topopyrones A–D (Figure 2) were discovered during research aimed at identifying new topoisomerase inhibitors [90,91]. Initial evidence [90,91] suggested that these compounds may be selective inhibitors of topoisomerase-I (topo-I), a potentially significant finding. It should be noted that two topoisomerases are known, namely topo-I and topo-II. These nuclear enzymes relax superhelical tension in DNA during replication, transcription and repair. They operate by reversibly breaking one (topo-I) or both (topo-II) strands in double-stranded DNA and unwinding the severed strand(s), thereby relieving torsional energy. Inhibition of topoisomerases, which are overexpressed in cancerous cells, is fatal to the cell [92]. Subsequent studies revealed that topopyrones are in fact dual inhibitors of topo-I and topo-II [93]: A finding that diminished the biomedical relevance of the natural products. Regardless, the biological properties of topopyrones are sufficiently interesting that a number of groups embarked on a total synthesis [94-96].
![[1860-5397-7-171-2]](/bjoc/content/figures/1860-5397-7-171-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our own involvement in this area was motivated by an interest in topoisomerase-I inhibitors, which are important antineoplastic resources [97], the archetype of which is camptothecin [81-83,98,99]. The objective of the present effort was to obtain the target molecules by any means, as rapidly as possible, and in a fashion that might enable the production of analogs for structure–activity relationship studies. By contrast, our work on fredericamycin and nothapodytine had chiefly reflected a desire to illustrate applications of the new methodology to the synthesis of interesting natural products.
The linearly fused topopyrones B and D are especially potent, and indeed, the activity of 53 against topo-I appears to be comparable to that of camptothecin [90,91,100,101]. Accordingly, our research centered on the linear series of compounds. A piece of information that was key to our retrosynthetic planning is that the action of alkali on 51 and 52 induces rearrangement to 53 and 54 [90,91], signifying that the linearly fused topopyrones are thermodynamically favored over their angular congeners. This implied that the cyclization of a precursor such as 55 under equilibrating conditions should selectively afford topopyrones B and D (Scheme 11). Drawing from the work of Snieckus [102,103], we further surmised that the assembly of 55 could be carried out in a single operation through the union of fragments 56 or 57 with aldehyde 58: The carrier of a moiety that is common to all topopyrones.
![[1860-5397-7-171-i11]](/bjoc/content/inline/1860-5397-7-171-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Retrosynthetic logic for the linear series of topopyrones.
Scheme 11: Retrosynthetic logic for the linear series of topopyrones.
A serviceable form of 58 proved to be compound 65, the preparation of which is outlined in Scheme 12. A key step in this sequence was the addition of the organolithium species 60, obtained through halogen–metal exchange of 59 with t-BuLi, to aldehyde 61 [104], leading to alcohol 62 in 67% yield. Straightforward manipulations of 62 then furnished the requisite 65.
![[1860-5397-7-171-i12]](/bjoc/content/inline/1860-5397-7-171-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Construction of the molecular subunit common to all topopyrones.
Scheme 12: Construction of the molecular subunit common to all topopyrones.
Interestingly, the 1H and 13C NMR spectra of all TIPS-protected compounds revealed that these substances exist as mixtures of atropdiastereomers. Furthermore, atropisomerism vanishes upon release of the TIPS unit (see below). An inspection of molecular models readily provides a qualitative illustration of this effect, in that the bulky TIPS group hampers rotation about the σ-bond connecting the benzylic carbon to the aryl segment (cf. arrows in 65). An MM+ conformational study [105] of a simplified analog of 63 estimated an energy barrier for internal rotation equal to about 17 kcal/mol [106].
As seen in Scheme 13, DAF technology came into the picture at the stage of the union of aldehyde 65 with amide 56. Thus, lithiation of benzamide 56 with sec-BuLi/TMEDA (1.05 equiv, 3 h, −78 °C) and addition of the resulting organolithium agent to 65 was presumed to form the alkoxide 66. In situ treatment of 66 with t-BuLi induced bromine–lithium exchange and cyclization of organolithium species 67 to a product that was believed to be 68. Aqueous workup arguably converted 68 into a dihydroanthraquinone, which upon exposure to the atmosphere was rapidly oxidized to the desired anthraquinone 69. The yield of this material was a modest 17% after purification, the remaining balance of the initial mass of 65 being the debrominated material 70 (60–70%). The NMR spectra of both products indicated that they existed as slowly interconverting atropdiastereomers, too. Numerous experiments aimed at improving the yield of 69 led to no fruitful outcome, revealing instead that the ratio of the two products remained essentially constant (ca. 1:3.5–4) regardless of the length of time (from 3 to 12 h) over which the reaction mixture was allowed to evolve following the addition of t-BuLi to 66.
![[1860-5397-7-171-i13]](/bjoc/content/inline/1860-5397-7-171-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Difficulties encountered during the merger of the topopyrone D moieties.
Scheme 13: Difficulties encountered during the merger of the topopyrone D moieties.
As a control experiment, we examined the preparation of the simpler anthraquinone 72 by the same method (Scheme 14). This exercise established that 72 formed considerably more efficiently than 69 (over 60% isolated yield), and that it was accompanied by only small amounts of the debrominated byproduct 73. We thus concluded that the low yield of 68 must have been the consequence of the consumption of a portion of aryllithium species 67 through parasitic proton-transfer steps, probably involving one of its benzylic positions as the proton donor. While the yield of 69 could not be improved, its preparation in the fashion just described is highly convergent. Furthermore, an overall yield around 20% for a one-pot sequence that involves three major steps (addition of lithiated 56 to 65, halogen–metal exchange, cyclization of aryllithium species 67) corresponds to an average of 55–60% yield per step. In such a light, the modest yield seemed quite acceptable, and indeed, with 69 in hand, the synthesis of topopyrone D was completed quickly.
![[1860-5397-7-171-i14]](/bjoc/content/inline/1860-5397-7-171-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Efficient synthesis of a simplified anthraquinone.
Scheme 14: Efficient synthesis of a simplified anthraquinone.
As shown in Scheme 15, desilylation of 69 (TBAF) furnished an alcohol that, contrary to the parent 69 or other TIPS-protected synthetic intermediates, exhibited no atropisomerism (single compound by 1H and 13C NMR). Oxidation (IBX) and treatment of the emerging ketone 74 with 48% aqueous HBr in AcOH under reflux afforded synthetic 54 in quantitative yield. No evidence for the formation of angular topopyrones could be garnered, reinforcing the notion that the linear series is the thermodynamically favored one. Substance 54 was fully characterized as such, as well as the considerably more soluble triacetyl derivative, as detailed in the isolation paper [90,91].
![[1860-5397-7-171-i15]](/bjoc/content/inline/1860-5397-7-171-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Total synthesis of topopyrone D.
Scheme 15: Total synthesis of topopyrone D.
An unexpected problem materialized when the foregoing sequence was transposed to the case of topopyrone B, the synthesis of which required the execution of the same operations starting with benzamide 57. Surprisingly, this material resisted deprotonation under Snieckus conditions. For instance, treatment with five equivalents (as opposed to the customary 1.05 equivalents) of sec-BuLi–TMEDA complex for a prolonged period of time resulted in no incorporation of deuterium upon quenching with CD3OD. The reasons for this remain unknown to this date. Various chlorinated benzamides undergo ortho-deprotonation without difficulty [104], implying that the resistance of 57 cannot be attributed to the chloro substituent per se. Nor can the problem be ascribed to sequestration of the base through coordination/chelation [33] effects involving the chlorine atom. Such a hypothesis fails to account for the fact that 57 resisted deprotonation even in the presence of excess base. Moreover, 2,3,4-trimethoxybenzamide, a congener of 57 in which an OMe group replaces the Cl substituent, undergoes ortho-metallation without incident [107], even though the triad of adjacent OMe groups can surely sequester organo–Li species at least as effectively as the 2,4-dimethoxy-3-chloro arrangement present in 57.
In any event, ortho-deprotonation was ultimately achieved by the reaction of 57 with the more basic t-BuLi–TMEDA complex (1 equiv, 3 h, –78 °C; complete deuterium incorporation upon CD3OD quench). Accordingly, the organolithium species thus generated was processed as detailed earlier in Scheme 14, leading to a mixture of the desired anthraquinone 76, obtained in 20% yield after chromatography, along with desbromo product 75, again isolated in 60–70% yield (Scheme 16). The chemistry leading to 76 thus performed just as efficiently (inefficiently?) as before. The elaboration of 77 to the poorly soluble topopyrone B proceeded uneventfully, and in accordance with the isolation paper [90,91]; the ultimate 53 was most readily characterized as the trimethyl ether [108].
![[1860-5397-7-171-i16]](/bjoc/content/inline/1860-5397-7-171-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Total synthesis of topopyrone B.
Scheme 16: Total synthesis of topopyrone B.
Conclusion
The work summarized in this review exemplifies, hopefully in a convincing fashion, the value of directed aromatic functionalization in the preparation of complex aromatic systems. Indeed, it is difficult today to imagine an efficient route to natural products such as fredericamycin, nothapoditine, and topopyrones that eschews DAF technology altogether. More importantly, the chemistry described herein is a testimony to the far-sightedness of the pioneers in the field of directed aromatic functionalization. These giants have provided the chemical community with immensely powerful tools that have truly revolutionized the business of producing elaborate aromatic compounds, both in the academic laboratory and in the industrial plant, and that continue to produce countless benefits to society in the form of new medicines and new materials.
References
-
Pandey, R. C.; Toussaint, M. W.; Stroshane, R. M.; Kalita, C. C.; Aszalos, A. A.; Garretson, A. L.; Wei, T. T.; Byrne, K. M.; Geoghegan, R. F., Jr.; White, R. J. J. Antibiot. 1981, 34, 1389.
Return to citation in text: [1] -
Warnick-Pickle, D. J.; Byrne, K. M.; Pandey, R. C.; White, R. J. J. Antibiot. 1981, 34, 1402.
Return to citation in text: [1] -
Misra, R.; Pandey, R. C.; Silverton, J. V. J. Am. Chem. Soc. 1982, 104, 4478. doi:10.1021/ja00380a025
Return to citation in text: [1] -
Misra, R.; Pandey, R. C.; Hilton, B. D.; Roller, P. P.; Silverton, J. V. J. Antibiot. 1987, 40, 786.
Return to citation in text: [1] -
Misra, R. J. Antibiot. 1988, 41, 976.
Return to citation in text: [1] -
Kelly, T. R.; Ohashi, N.; Armstrong-Chong, R. J.; Bell, S. H. J. Am. Chem. Soc. 1986, 108, 7100. doi:10.1021/ja00282a042
Return to citation in text: [1] [2] [3] [4] -
Kelly, T. R.; Bell, S. H.; Ohashi, N.; Armstrong-Chong, R. J. J. Am. Chem. Soc. 1988, 110, 6471. doi:10.1021/ja00227a030
Return to citation in text: [1] [2] [3] [4] -
Clive, D. L. J.; Tao, Y.; Khodabocus, A.; Wu, Y. J.; Angoh, A. G.; Bennett, S. M.; Boddy, C. N.; Bordeleau, L.; Kellner, D.; Kleiner, G.; Middleton, D. S.; Nichols, C. J.; Richardson, S. R.; Vernon, P. G. J. Chem. Soc., Chem. Commun. 1992, 1489. doi:10.1039/C39920001489
Return to citation in text: [1] -
Rama Rao, A. V.; Singh, A. K.; Rao, B. V.; Reddy, K. M. Tetrahedron Lett. 1993, 34, 2665. doi:10.1016/S0040-4039(00)77651-9
Return to citation in text: [1] -
Saint-Jalmes, L.; Lila, C.; Xu, J. Z.; Moreau, L.; Pfeiffer, B.; Eck, G.; Pelsez, L.; Rolando, C.; Julia, M. Bull. Soc. Chim. Fr. 1993, 130, 447.
Return to citation in text: [1] -
Wendt, J. A.; Gauvreau, P. J.; Bach, R. D. J. Am. Chem. Soc. 1994, 116, 9921. doi:10.1021/ja00101a013
Return to citation in text: [1] -
Rama Rao, A. V.; Singh, A. K.; Rao, B. V.; Reddy, K. M. Heterocycles 1994, 37, 1893. doi:10.3987/COM-93-S163
Return to citation in text: [1] -
Clive, D. L. J.; Tao, Y.; Khodabocus, A.; Wu, Y.-J.; Angoh, A. G.; Bennett, S. M.; Boddy, C. N.; Bordeleau, L.; Kellner, D.; Kleiner, G.; Middleton, D. S.; Nichols, C. J.; Richardson, S. R.; Vernon, P. G. J. Am. Chem. Soc. 1994, 116, 11275. doi:10.1021/ja00104a009
Return to citation in text: [1] -
Boger, D. L.; Hueter, O.; Mbiya, K.; Zhang, M. J. Am. Chem. Soc. 1995, 117, 11839. doi:10.1021/ja00153a004
Return to citation in text: [1] -
Kita, Y.; Higuchi, K.; Yoshida, Y.; Iio, K.; Kitagaki, S.; Akai, S.; Fujioka, H. Angew. Chem., Int. Ed. 1999, 38, 683. doi:10.1002/(SICI)1521-3773(19990301)38:5<683::AID-ANIE683>3.0.CO;2-0
Return to citation in text: [1] -
Kita, Y.; Iio, K.; Kawaguchi, K.-I.; Fukuda, N.; Takeda, Y.; Ueno, H.; Okunaka, R.; Higuchi, K.; Tsujino, T.; Fujioka, H.; Akai, S. Chem.–Eur. J. 2000, 6, 3897. doi:10.1002/1521-3765(20001103)6:21<3897::AID-CHEM3897>3.0.CO;2-1
Return to citation in text: [1] -
Most of the authors cited above described extensive exploratory work before completing a total synthesis. For a complete bibliography of such background studies, see the literature cited therein.
Return to citation in text: [1] -
Parker, K. A.; Koziski, K. A.; Breault, G. Tetrahedron Lett. 1985, 26, 2181. doi:10.1016/S0040-4039(00)98956-1
Return to citation in text: [1] -
Kende, A. S.; Ebetino, F. H.; Ohta, T. Tetrahedron Lett. 1985, 26, 3063. doi:10.1016/S0040-4039(00)98619-2
Return to citation in text: [1] -
Braun, M.; Veith, R. Tetrahedron Lett. 1986, 27, 179. doi:10.1016/S0040-4039(00)83971-4
Return to citation in text: [1] -
Parker, K. A.; Breault, G. A. Tetrahedron Lett. 1986, 27, 3835. doi:10.1016/S0040-4039(00)83892-7
Return to citation in text: [1] -
Mehta, G.; Subrahmanyam, D. Tetrahedron Lett. 1987, 28, 479. doi:10.1016/S0040-4039(00)95761-7
Return to citation in text: [1] -
Naik, S. N.; Pandey, B.; Ayyangar, N. R. Synth. Commun. 1988, 18, 633. doi:10.1080/00397918808064022
Return to citation in text: [1] -
Toyota, M.; Terashima, S. Tetrahedron Lett. 1989, 30, 829. doi:10.1016/S0040-4039(01)80627-4
Return to citation in text: [1] -
Aidhen, I. S.; Narasimhan, N. S. Tetrahedron Lett. 1989, 30, 5323. doi:10.1016/S0040-4039(01)93776-1
Return to citation in text: [1] -
Pandey, B.; Khire, U. R.; Ayyangar, N. R. J. Chem. Soc., Chem. Commun. 1990, 1791. doi:10.1039/C39900001791
Return to citation in text: [1] -
Watanabe, M.; Morimoto, H.; Furukawa, S. Heterocycles 1993, 36, 2681. doi:10.3987/COM-93-6539
Return to citation in text: [1] -
Kessar, S. V.; Vohra, R.; Kaur, N. P.; Singh, K. N.; Singh, P. Chem. Commun. 1994, 1327. doi:10.1039/C39940001327
Return to citation in text: [1] -
Evans, P. A.; Brandt, T. A. Tetrahedron Lett. 1996, 37, 1367. doi:10.1016/0040-4039(96)00055-X
Return to citation in text: [1] -
Baskaran, S.; Nagy, E.; Braun, M. Liebigs Ann. 1997, 311. doi:10.1002/jlac.199719970206
Return to citation in text: [1] -
Browne, M. E. Synthetic Studies Toward an Advanced Intermediate of Fredericamycin A and the Development of a Novel Palladium(0)-Mediated Spiroarylation. Ph.D. Thesis, Rice University, Houston, TX, 1991.
Return to citation in text: [1] [2] -
Snieckus, V. Chem. Rev. 1990, 90, 879. doi:10.1021/cr00104a001
Return to citation in text: [1] -
Hartung, C. G.; Snieckus, V. Chapter 10. The Directed ortho Metalation Reaction – A Point of Departure for New Synthetic Aromatic Chemistry. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH Verlag GmbH & KGaA: Weinheim, Germany, 2002; pp 330–367. doi:10.1002/3527601767.ch10
Return to citation in text: [1] [2] -
Whisler, M.; MacNeil, S.; Snieckus, V.; Beak, P. Angew. Chem., Int. Ed. 2004, 43, 2206–2225. doi:10.1002/anie.200300590
Return to citation in text: [1] -
Macklin, T.; Snieckus, V. In Handbook of C-H Tansformations; Dyker, G., Ed.; Wiley-VCH: New York, 2005; pp 106–119.
Return to citation in text: [1] -
Poindexter, G. S. J. Org. Chem. 1982, 47, 3787. doi:10.1021/jo00140a047
Return to citation in text: [1] -
Horiuchi, C. A.; Satoh, J. Y. Bull. Chem. Soc. Jpn. 1984, 57, 2691. doi:10.1246/bcsj.57.2691
Return to citation in text: [1] -
Ciufolini, M. A.; Browne, M. E. Tetrahedron Lett. 1987, 28, 171. doi:10.1016/S0040-4039(00)95678-8
Return to citation in text: [1] -
Ciufolini, M. A.; Qi, H. B.; Browne, M. E. J. Org. Chem. 1988, 53, 4149. doi:10.1021/jo00252a064
Return to citation in text: [1] -
Moradi, W. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 7996. doi:10.1021/ja010797+
Return to citation in text: [1] -
Hamada, T.; Chieffi, A.; Åhman, J.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 1261. doi:10.1021/ja011122+
Return to citation in text: [1] -
Hamada, T.; Buchwald, S. L. Org. Lett. 2002, 4, 999. doi:10.1021/ol025563p
Return to citation in text: [1] -
Nguyen, H. N.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11818. doi:10.1021/ja036947t
Return to citation in text: [1] -
Hartwig, J. F. Synlett 2006, 1283. doi:10.1055/s-2006-939728
Return to citation in text: [1] -
Liao, X.; Weng, Z.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 195. doi:10.1021/ja074453g
Return to citation in text: [1] -
Hama, T.; Hartwig, J. F. Org. Lett. 2008, 10, 1549. doi:10.1021/ol800258u
Return to citation in text: [1] -
Ciufolini, M. A.; Wood, C. Y. Tetrahedron Lett. 1986, 27, 5085. doi:10.1016/S0040-4039(00)85139-4
Return to citation in text: [1] -
Drueckhammer, D. G.; Barbas, C. F., III; Nozaki, K.; Wong, C. H.; Wood, C. Y.; Ciufolini, M. A. J. Org. Chem. 1988, 53, 1607. doi:10.1021/jo00243a003
Return to citation in text: [1] -
Ciufolini, M. A.; Hermann, C. W.; Whitmire, K. H.; Byrne, N. E. J. Am. Chem. Soc. 1989, 111, 3473. doi:10.1021/ja00191a078
Return to citation in text: [1] -
Ciufolini, M. A.; Dong, Q. Chem. Commun. 1996, 881. doi:10.1039/cc9960000881
Return to citation in text: [1] -
Ciufolini, M. A.; Shimizu, T.; Swaminathan, S.; Xi, N. Tetrahedron Lett. 1997, 38, 4947. doi:10.1016/S0040-4039(97)01087-3
Return to citation in text: [1] -
Ciufolini, M. A.; Hermann, C. Y. W.; Dong, Q.; Shimizu, T.; Swaminathan, S.; Xi, N. Synlett 1998, 105. doi:10.1055/s-1998-1584
Return to citation in text: [1] -
Cassidy, M. P.; Padwa, A. Org. Lett. 2004, 6, 4029. doi:10.1021/ol048326q
Return to citation in text: [1] -
Leverett, C. A.; Cassidy, M. P.; Padwa, A. J. Org. Chem. 2006, 71, 8591. doi:10.1021/jo0616714
Return to citation in text: [1] -
Kiren, S.; Hong, X.; Leverett, C. A.; Padwa, A. Tetrahedron 2009, 65, 6720. doi:10.1016/j.tet.2009.03.011
Return to citation in text: [1] -
Padwa, A.; Leverett, C. A.; Hong, X. Acta Chim. Slov. 2009, 56, 527.
Return to citation in text: [1] -
Zhou, W.-S.; Lu, Z.-H.; Wang, Z.-M. Tetrahedron Lett. 1991, 32, 1467. doi:10.1016/0040-4039(91)80360-I
Return to citation in text: [1] -
Xu, Y.-M.; Zhou, W.-S. J. Chem. Soc., Perkin Trans. 1 1997, 741. doi:10.1039/A605128F
Return to citation in text: [1] -
Yang, C.; Liao, L.; Xu, Y.; Zhang, H.; Xia, P.; Zhou, W. Tetrahedron: Asymmetry 1999, 10, 2311. doi:10.1016/S0957-4166(99)00239-6
Return to citation in text: [1] -
Liao, L.-X.; Wang, Z.-M.; Zhang, H.-X.; Zhou, W.-S. Tetrahedron: Asymmetry 1999, 10, 3649. doi:10.1016/S0957-4166(99)00382-1
Return to citation in text: [1] -
Haukaas, M. H.; O'Doherty, G. A. Org. Lett. 2001, 3, 401. doi:10.1021/ol006907j
Return to citation in text: [1] -
Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2004, 126, 11804. doi:10.1021/ja046185h
Return to citation in text: [1] -
Wu, T.-S.; Chan, Y.-Y.; Leu, Y.-L.; Chern, C.-Y.; Chen, C.-F. Phytochemistry 1996, 42, 907. doi:10.1016/0031-9422(95)00962-0
Return to citation in text: [1] -
Govindachari, T. R.; Ravindranath, K. R.; Viswanathan, N. J. Chem. Soc., Perkin Trans. 1 1974, 1215. doi:10.1039/p19740001215
Return to citation in text: [1] -
Kametani, T.; Takeda, H.; Nemoto, H.; Fukumoto, K. J. Chem. Soc., Perkin Trans. 1 1975, 1825.
Return to citation in text: [1] -
Pendrak, I.; Barney, S.; Wittrock, R.; Lambert, D. M.; Kingsbury, W. D. J. Org. Chem. 1994, 59, 2623. doi:10.1021/jo00088a057
Return to citation in text: [1] -
Pendrak, I.; Wittrock, R.; Kingsbury, W. D. J. Org. Chem. 1995, 60, 2912. doi:10.1021/jo00114a050
Return to citation in text: [1] -
Boger, D. L.; Hong, J. J. Am. Chem. Soc. 1998, 120, 1218. doi:10.1021/ja973007y
Return to citation in text: [1] [2] -
Boger, D. L. J. Heterocycl. Chem. 1998, 35, 1003. doi:10.1002/jhet.5570350502
Return to citation in text: [1] [2] -
Bowman, W. R.; Bridge, C. F.; Brookes, P.; Cloonan, M. O.; Leach, D. C. J. Chem. Soc., Perkin Trans. 1 2002, 58.
Return to citation in text: [1] -
Kato, I.; Higashimoto, M.; Tamura, O.; Ishibashi, H. J. Org. Chem. 2003, 68, 7983. doi:10.1021/jo030177m
Return to citation in text: [1] -
Raolji, G. B.; Garçon, S.; Greene, A. E.; Kanazawa, A. Angew. Chem., Int. Ed. 2003, 42, 5059. doi:10.1002/anie.200352094
Return to citation in text: [1] -
Chavan, S. P.; Sivappa, R. Tetrahedron Lett. 2004, 45, 3941. doi:10.1016/j.tetlet.2004.03.089
Return to citation in text: [1] -
Bowman, W. R.; Cloonan, M. O.; Fletcher, A. J.; Stein, T. Org. Biomol. Chem. 2005, 3, 1460. doi:10.1039/b501509j
Return to citation in text: [1] -
Toyota, M.; Komori, C.; Ihara, M. Heterocycles 2000, 52, 591. doi:10.3987/COM-99-S109
Return to citation in text: [1] -
Lin, C. H.; Tsai, M. R.; Wang, Y. S.; Chang, N. C. J. Org. Chem. 2003, 68, 5688. doi:10.1021/jo034042s
Return to citation in text: [1] -
Mekouar, K.; Génisson, Y.; Leue, S.; Greene, A. E. J. Org. Chem. 2000, 65, 5212. doi:10.1021/jo0003448
Return to citation in text: [1] -
Toyota, M.; Komori, C.; Ihara, M. J. Org. Chem. 2000, 65, 7110. doi:10.1021/jo000816i
Return to citation in text: [1] -
Bowman, W. R.; Bridge, C. F.; Cloonan, M. O.; Leach, D. C. Synlett 2001, 765. doi:10.1055/s-2001-14592
Return to citation in text: [1] -
Ciufolini, M. A.; Chan, B. K. Heterocycles 2007, 74, 101. doi:10.3987/REV-07-SR(W)4
See for a review.
Return to citation in text: [1] -
Ciufolini, M. A.; Roschangar, F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1692. doi:10.1002/anie.199616921
Return to citation in text: [1] [2] [3] -
Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049. doi:10.1016/S0040-4020(97)00365-7
Return to citation in text: [1] [2] [3] -
Ciufolini, M. A.; Roschangar, F. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2000; Vol. 4.
Return to citation in text: [1] [2] [3] -
The Boger synthesis of 36 had earlier validated this approach.
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Ciufolini, M. A.; Mitchell, J. W.; Roschangar, F. Tetrahedron Lett. 1996, 37, 8281. doi:10.1016/0040-4039(96)01937-5
Return to citation in text: [1] -
Thompson, W. J.; Gaudino, J. J. Org. Chem. 1984, 49, 5237. doi:10.1021/jo00200a045
Return to citation in text: [1] -
Kobayashi, Y.; Nakano, M.; Kumar, G. B.; Kishihara, K. J. Org. Chem. 1998, 63, 7505. doi:10.1021/jo980942a
Return to citation in text: [1] -
Carles, L.; Narkunan, K.; Penlou, S.; Rousset, L.; Bouchu, D.; Ciufolini, M. A. J. Org. Chem. 2002, 67, 4304. doi:10.1021/jo025546d
Return to citation in text: [1] -
Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Champoux, J. J. Annu. Rev. Biochem. 2001, 70, 369. doi:10.1146/annurev.biochem.70.1.369
Return to citation in text: [1] -
Khan, Q. A.; Elban, M. A.; Hecht, S. M. J. Am. Chem. Soc. 2008, 130, 12888. doi:10.1021/ja805421b
Return to citation in text: [1] -
Qi, L. Study Directed to the Total Synthesis of Kidamycin and Topopyrones B and D. Ph.D. Thesis, Brown University, Providence, RI, 2003.
Return to citation in text: [1] -
Gattinoni, S.; Merlini, L.; Dallavalle, S. Tetrahedron Lett. 2007, 48, 1049. doi:10.1016/j.tetlet.2006.11.164
Return to citation in text: [1] -
Elban, M. A.; Hecht, S. M. J. Org. Chem. 2008, 73, 785. doi:10.1021/jo702487r
Return to citation in text: [1] -
Denny, W. A. Expert Opin. Emerging Drugs 2004, 9, 105. doi:10.1517/14728214.9.1.105
Return to citation in text: [1] -
Rothenberg, M. L. Ann. Oncol. 1997, 8, 837. doi:10.1023/A:1008270717294
Return to citation in text: [1] -
Versace, R. W. Expert Opin. Ther. Pat. 2003, 13, 751. doi:10.1517/13543776.13.6.751
Return to citation in text: [1] -
Dallavalle, S.; Gattinoni, S.; Mazzini, S.; Scaglioni, L.; Merlini, L.; Tinelli, S.; Beretta, G. L.; Zunino, F. Bioorg. Med. Chem. Lett. 2008, 18, 1484. doi:10.1016/j.bmcl.2007.12.055
Return to citation in text: [1] -
Scaglioni, L.; Mazzini, S.; Mondelli, R.; Dallavalle, S.; Gattinoni, S.; Tinelli, S.; Beretta, G. L.; Zunino, F.; Ragg, E. Bioorg. Med. Chem. 2009, 17, 484. doi:10.1016/j.bmc.2008.12.005
Return to citation in text: [1] -
Wang, X.; Snieckus, V. Synlett 1990, 313. doi:10.1055/s-1990-21074
Return to citation in text: [1] -
De Silva, S. O.; Watanabe, M.; Snieckus, V. J. Org. Chem. 1979, 44, 4802. doi:10.1021/jo00394a012
Return to citation in text: [1] -
Langer, P.; Freifeld, I. Synlett 2001, 4, 523. doi:10.1055/s-2001-12327
Return to citation in text: [1] [2] -
Calculations were carried out with the Hyperchem(TM) package.
Return to citation in text: [1] -
Rotational barriers are notoriously difficult to estimate, at least by MM+, and the values thus found are often lower than in reality. We thus presume that the calculated value represents a lower limit for the size of the rotational barrier in question.
Return to citation in text: [1] -
Sibi, M. P.; Jalil Miah, M. A.; Snieckus, V. J. Org. Chem. 1984, 49, 737. doi:10.1021/jo00179a001
Return to citation in text: [1] -
Tan, J. S.; Ciufolini, M. A. Org. Lett. 2006, 8, 4771. doi:10.1021/ol0617291
Return to citation in text: [1]
| 86. | Ciufolini, M. A.; Mitchell, J. W.; Roschangar, F. Tetrahedron Lett. 1996, 37, 8281. doi:10.1016/0040-4039(96)01937-5 |
| 87. | Thompson, W. J.; Gaudino, J. J. Org. Chem. 1984, 49, 5237. doi:10.1021/jo00200a045 |
| 81. | Ciufolini, M. A.; Roschangar, F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1692. doi:10.1002/anie.199616921 |
| 82. | Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049. doi:10.1016/S0040-4020(97)00365-7 |
| 83. | Ciufolini, M. A.; Roschangar, F. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2000; Vol. 4. |
| 93. | Khan, Q. A.; Elban, M. A.; Hecht, S. M. J. Am. Chem. Soc. 2008, 130, 12888. doi:10.1021/ja805421b |
| 94. | Qi, L. Study Directed to the Total Synthesis of Kidamycin and Topopyrones B and D. Ph.D. Thesis, Brown University, Providence, RI, 2003. |
| 95. | Gattinoni, S.; Merlini, L.; Dallavalle, S. Tetrahedron Lett. 2007, 48, 1049. doi:10.1016/j.tetlet.2006.11.164 |
| 96. | Elban, M. A.; Hecht, S. M. J. Org. Chem. 2008, 73, 785. doi:10.1021/jo702487r |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 92. | Champoux, J. J. Annu. Rev. Biochem. 2001, 70, 369. doi:10.1146/annurev.biochem.70.1.369 |
| 68. | Boger, D. L.; Hong, J. J. Am. Chem. Soc. 1998, 120, 1218. doi:10.1021/ja973007y |
| 69. | Boger, D. L. J. Heterocycl. Chem. 1998, 35, 1003. doi:10.1002/jhet.5570350502 |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 88. | Kobayashi, Y.; Nakano, M.; Kumar, G. B.; Kishihara, K. J. Org. Chem. 1998, 63, 7505. doi:10.1021/jo980942a |
| 89. | Carles, L.; Narkunan, K.; Penlou, S.; Rousset, L.; Bouchu, D.; Ciufolini, M. A. J. Org. Chem. 2002, 67, 4304. doi:10.1021/jo025546d |
| 97. | Denny, W. A. Expert Opin. Emerging Drugs 2004, 9, 105. doi:10.1517/14728214.9.1.105 |
| 81. | Ciufolini, M. A.; Roschangar, F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1692. doi:10.1002/anie.199616921 |
| 82. | Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049. doi:10.1016/S0040-4020(97)00365-7 |
| 83. | Ciufolini, M. A.; Roschangar, F. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2000; Vol. 4. |
| 98. | Rothenberg, M. L. Ann. Oncol. 1997, 8, 837. doi:10.1023/A:1008270717294 |
| 99. | Versace, R. W. Expert Opin. Ther. Pat. 2003, 13, 751. doi:10.1517/13543776.13.6.751 |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 100. | Dallavalle, S.; Gattinoni, S.; Mazzini, S.; Scaglioni, L.; Merlini, L.; Tinelli, S.; Beretta, G. L.; Zunino, F. Bioorg. Med. Chem. Lett. 2008, 18, 1484. doi:10.1016/j.bmcl.2007.12.055 |
| 101. | Scaglioni, L.; Mazzini, S.; Mondelli, R.; Dallavalle, S.; Gattinoni, S.; Tinelli, S.; Beretta, G. L.; Zunino, F.; Ragg, E. Bioorg. Med. Chem. 2009, 17, 484. doi:10.1016/j.bmc.2008.12.005 |
| 33. | Hartung, C. G.; Snieckus, V. Chapter 10. The Directed ortho Metalation Reaction – A Point of Departure for New Synthetic Aromatic Chemistry. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH Verlag GmbH & KGaA: Weinheim, Germany, 2002; pp 330–367. doi:10.1002/3527601767.ch10 |
| 106. | Rotational barriers are notoriously difficult to estimate, at least by MM+, and the values thus found are often lower than in reality. We thus presume that the calculated value represents a lower limit for the size of the rotational barrier in question. |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 102. | Wang, X.; Snieckus, V. Synlett 1990, 313. doi:10.1055/s-1990-21074 |
| 103. | De Silva, S. O.; Watanabe, M.; Snieckus, V. J. Org. Chem. 1979, 44, 4802. doi:10.1021/jo00394a012 |
| 90. | Kanai, Y.; Ishiyama, D.; Senda, H.; Iwatani, W.; Takahaski, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. J. Antibiot. 2000, 53, 863. |
| 91. | Ishiyama, D.; Kanai, Y.; Senda, H.; Iwatani, W.; Takahashi, H.; Konno, H.; Kanazawa, S. J. Antibiot. 2000, 53, 873. |
| 108. | Tan, J. S.; Ciufolini, M. A. Org. Lett. 2006, 8, 4771. doi:10.1021/ol0617291 |
| 107. | Sibi, M. P.; Jalil Miah, M. A.; Snieckus, V. J. Org. Chem. 1984, 49, 737. doi:10.1021/jo00179a001 |
| 1. | Pandey, R. C.; Toussaint, M. W.; Stroshane, R. M.; Kalita, C. C.; Aszalos, A. A.; Garretson, A. L.; Wei, T. T.; Byrne, K. M.; Geoghegan, R. F., Jr.; White, R. J. J. Antibiot. 1981, 34, 1389. |
| 2. | Warnick-Pickle, D. J.; Byrne, K. M.; Pandey, R. C.; White, R. J. J. Antibiot. 1981, 34, 1402. |
| 3. | Misra, R.; Pandey, R. C.; Silverton, J. V. J. Am. Chem. Soc. 1982, 104, 4478. doi:10.1021/ja00380a025 |
| 4. | Misra, R.; Pandey, R. C.; Hilton, B. D.; Roller, P. P.; Silverton, J. V. J. Antibiot. 1987, 40, 786. |
| 5. | Misra, R. J. Antibiot. 1988, 41, 976. |
| 32. | Snieckus, V. Chem. Rev. 1990, 90, 879. doi:10.1021/cr00104a001 |
| 33. | Hartung, C. G.; Snieckus, V. Chapter 10. The Directed ortho Metalation Reaction – A Point of Departure for New Synthetic Aromatic Chemistry. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH Verlag GmbH & KGaA: Weinheim, Germany, 2002; pp 330–367. doi:10.1002/3527601767.ch10 |
| 34. | Whisler, M.; MacNeil, S.; Snieckus, V.; Beak, P. Angew. Chem., Int. Ed. 2004, 43, 2206–2225. doi:10.1002/anie.200300590 |
| 35. | Macklin, T.; Snieckus, V. In Handbook of C-H Tansformations; Dyker, G., Ed.; Wiley-VCH: New York, 2005; pp 106–119. |
| 44. | Hartwig, J. F. Synlett 2006, 1283. doi:10.1055/s-2006-939728 |
| 45. | Liao, X.; Weng, Z.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 195. doi:10.1021/ja074453g |
| 46. | Hama, T.; Hartwig, J. F. Org. Lett. 2008, 10, 1549. doi:10.1021/ol800258u |
| 31. | Browne, M. E. Synthetic Studies Toward an Advanced Intermediate of Fredericamycin A and the Development of a Novel Palladium(0)-Mediated Spiroarylation. Ph.D. Thesis, Rice University, Houston, TX, 1991. |
| 47. | Ciufolini, M. A.; Wood, C. Y. Tetrahedron Lett. 1986, 27, 5085. doi:10.1016/S0040-4039(00)85139-4 |
| 48. | Drueckhammer, D. G.; Barbas, C. F., III; Nozaki, K.; Wong, C. H.; Wood, C. Y.; Ciufolini, M. A. J. Org. Chem. 1988, 53, 1607. doi:10.1021/jo00243a003 |
| 49. | Ciufolini, M. A.; Hermann, C. W.; Whitmire, K. H.; Byrne, N. E. J. Am. Chem. Soc. 1989, 111, 3473. doi:10.1021/ja00191a078 |
| 50. | Ciufolini, M. A.; Dong, Q. Chem. Commun. 1996, 881. doi:10.1039/cc9960000881 |
| 51. | Ciufolini, M. A.; Shimizu, T.; Swaminathan, S.; Xi, N. Tetrahedron Lett. 1997, 38, 4947. doi:10.1016/S0040-4039(97)01087-3 |
| 52. | Ciufolini, M. A.; Hermann, C. Y. W.; Dong, Q.; Shimizu, T.; Swaminathan, S.; Xi, N. Synlett 1998, 105. doi:10.1055/s-1998-1584 |
| 17. | Most of the authors cited above described extensive exploratory work before completing a total synthesis. For a complete bibliography of such background studies, see the literature cited therein. |
| 18. | Parker, K. A.; Koziski, K. A.; Breault, G. Tetrahedron Lett. 1985, 26, 2181. doi:10.1016/S0040-4039(00)98956-1 |
| 19. | Kende, A. S.; Ebetino, F. H.; Ohta, T. Tetrahedron Lett. 1985, 26, 3063. doi:10.1016/S0040-4039(00)98619-2 |
| 20. | Braun, M.; Veith, R. Tetrahedron Lett. 1986, 27, 179. doi:10.1016/S0040-4039(00)83971-4 |
| 21. | Parker, K. A.; Breault, G. A. Tetrahedron Lett. 1986, 27, 3835. doi:10.1016/S0040-4039(00)83892-7 |
| 22. | Mehta, G.; Subrahmanyam, D. Tetrahedron Lett. 1987, 28, 479. doi:10.1016/S0040-4039(00)95761-7 |
| 23. | Naik, S. N.; Pandey, B.; Ayyangar, N. R. Synth. Commun. 1988, 18, 633. doi:10.1080/00397918808064022 |
| 24. | Toyota, M.; Terashima, S. Tetrahedron Lett. 1989, 30, 829. doi:10.1016/S0040-4039(01)80627-4 |
| 25. | Aidhen, I. S.; Narasimhan, N. S. Tetrahedron Lett. 1989, 30, 5323. doi:10.1016/S0040-4039(01)93776-1 |
| 26. | Pandey, B.; Khire, U. R.; Ayyangar, N. R. J. Chem. Soc., Chem. Commun. 1990, 1791. doi:10.1039/C39900001791 |
| 27. | Watanabe, M.; Morimoto, H.; Furukawa, S. Heterocycles 1993, 36, 2681. doi:10.3987/COM-93-6539 |
| 28. | Kessar, S. V.; Vohra, R.; Kaur, N. P.; Singh, K. N.; Singh, P. Chem. Commun. 1994, 1327. doi:10.1039/C39940001327 |
| 29. | Evans, P. A.; Brandt, T. A. Tetrahedron Lett. 1996, 37, 1367. doi:10.1016/0040-4039(96)00055-X |
| 30. | Baskaran, S.; Nagy, E.; Braun, M. Liebigs Ann. 1997, 311. doi:10.1002/jlac.199719970206 |
| 31. | Browne, M. E. Synthetic Studies Toward an Advanced Intermediate of Fredericamycin A and the Development of a Novel Palladium(0)-Mediated Spiroarylation. Ph.D. Thesis, Rice University, Houston, TX, 1991. |
| 6. | Kelly, T. R.; Ohashi, N.; Armstrong-Chong, R. J.; Bell, S. H. J. Am. Chem. Soc. 1986, 108, 7100. doi:10.1021/ja00282a042 |
| 7. | Kelly, T. R.; Bell, S. H.; Ohashi, N.; Armstrong-Chong, R. J. J. Am. Chem. Soc. 1988, 110, 6471. doi:10.1021/ja00227a030 |
| 8. | Clive, D. L. J.; Tao, Y.; Khodabocus, A.; Wu, Y. J.; Angoh, A. G.; Bennett, S. M.; Boddy, C. N.; Bordeleau, L.; Kellner, D.; Kleiner, G.; Middleton, D. S.; Nichols, C. J.; Richardson, S. R.; Vernon, P. G. J. Chem. Soc., Chem. Commun. 1992, 1489. doi:10.1039/C39920001489 |
| 9. | Rama Rao, A. V.; Singh, A. K.; Rao, B. V.; Reddy, K. M. Tetrahedron Lett. 1993, 34, 2665. doi:10.1016/S0040-4039(00)77651-9 |
| 10. | Saint-Jalmes, L.; Lila, C.; Xu, J. Z.; Moreau, L.; Pfeiffer, B.; Eck, G.; Pelsez, L.; Rolando, C.; Julia, M. Bull. Soc. Chim. Fr. 1993, 130, 447. |
| 11. | Wendt, J. A.; Gauvreau, P. J.; Bach, R. D. J. Am. Chem. Soc. 1994, 116, 9921. doi:10.1021/ja00101a013 |
| 12. | Rama Rao, A. V.; Singh, A. K.; Rao, B. V.; Reddy, K. M. Heterocycles 1994, 37, 1893. doi:10.3987/COM-93-S163 |
| 13. | Clive, D. L. J.; Tao, Y.; Khodabocus, A.; Wu, Y.-J.; Angoh, A. G.; Bennett, S. M.; Boddy, C. N.; Bordeleau, L.; Kellner, D.; Kleiner, G.; Middleton, D. S.; Nichols, C. J.; Richardson, S. R.; Vernon, P. G. J. Am. Chem. Soc. 1994, 116, 11275. doi:10.1021/ja00104a009 |
| 14. | Boger, D. L.; Hueter, O.; Mbiya, K.; Zhang, M. J. Am. Chem. Soc. 1995, 117, 11839. doi:10.1021/ja00153a004 |
| 15. | Kita, Y.; Higuchi, K.; Yoshida, Y.; Iio, K.; Kitagaki, S.; Akai, S.; Fujioka, H. Angew. Chem., Int. Ed. 1999, 38, 683. doi:10.1002/(SICI)1521-3773(19990301)38:5<683::AID-ANIE683>3.0.CO;2-0 |
| 16. | Kita, Y.; Iio, K.; Kawaguchi, K.-I.; Fukuda, N.; Takeda, Y.; Ueno, H.; Okunaka, R.; Higuchi, K.; Tsujino, T.; Fujioka, H.; Akai, S. Chem.–Eur. J. 2000, 6, 3897. doi:10.1002/1521-3765(20001103)6:21<3897::AID-CHEM3897>3.0.CO;2-1 |
| 40. | Moradi, W. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 7996. doi:10.1021/ja010797+ |
| 41. | Hamada, T.; Chieffi, A.; Åhman, J.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 1261. doi:10.1021/ja011122+ |
| 42. | Hamada, T.; Buchwald, S. L. Org. Lett. 2002, 4, 999. doi:10.1021/ol025563p |
| 43. | Nguyen, H. N.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11818. doi:10.1021/ja036947t |
| 6. | Kelly, T. R.; Ohashi, N.; Armstrong-Chong, R. J.; Bell, S. H. J. Am. Chem. Soc. 1986, 108, 7100. doi:10.1021/ja00282a042 |
| 7. | Kelly, T. R.; Bell, S. H.; Ohashi, N.; Armstrong-Chong, R. J. J. Am. Chem. Soc. 1988, 110, 6471. doi:10.1021/ja00227a030 |
| 39. | Ciufolini, M. A.; Qi, H. B.; Browne, M. E. J. Org. Chem. 1988, 53, 4149. doi:10.1021/jo00252a064 |
| 37. | Horiuchi, C. A.; Satoh, J. Y. Bull. Chem. Soc. Jpn. 1984, 57, 2691. doi:10.1246/bcsj.57.2691 |
| 6. | Kelly, T. R.; Ohashi, N.; Armstrong-Chong, R. J.; Bell, S. H. J. Am. Chem. Soc. 1986, 108, 7100. doi:10.1021/ja00282a042 |
| 7. | Kelly, T. R.; Bell, S. H.; Ohashi, N.; Armstrong-Chong, R. J. J. Am. Chem. Soc. 1988, 110, 6471. doi:10.1021/ja00227a030 |
| 6. | Kelly, T. R.; Ohashi, N.; Armstrong-Chong, R. J.; Bell, S. H. J. Am. Chem. Soc. 1986, 108, 7100. doi:10.1021/ja00282a042 |
| 7. | Kelly, T. R.; Bell, S. H.; Ohashi, N.; Armstrong-Chong, R. J. J. Am. Chem. Soc. 1988, 110, 6471. doi:10.1021/ja00227a030 |
| 38. | Ciufolini, M. A.; Browne, M. E. Tetrahedron Lett. 1987, 28, 171. doi:10.1016/S0040-4039(00)95678-8 |
| 64. | Govindachari, T. R.; Ravindranath, K. R.; Viswanathan, N. J. Chem. Soc., Perkin Trans. 1 1974, 1215. doi:10.1039/p19740001215 |
| 53. | Cassidy, M. P.; Padwa, A. Org. Lett. 2004, 6, 4029. doi:10.1021/ol048326q |
| 54. | Leverett, C. A.; Cassidy, M. P.; Padwa, A. J. Org. Chem. 2006, 71, 8591. doi:10.1021/jo0616714 |
| 55. | Kiren, S.; Hong, X.; Leverett, C. A.; Padwa, A. Tetrahedron 2009, 65, 6720. doi:10.1016/j.tet.2009.03.011 |
| 56. | Padwa, A.; Leverett, C. A.; Hong, X. Acta Chim. Slov. 2009, 56, 527. |
| 57. | Zhou, W.-S.; Lu, Z.-H.; Wang, Z.-M. Tetrahedron Lett. 1991, 32, 1467. doi:10.1016/0040-4039(91)80360-I |
| 58. | Xu, Y.-M.; Zhou, W.-S. J. Chem. Soc., Perkin Trans. 1 1997, 741. doi:10.1039/A605128F |
| 59. | Yang, C.; Liao, L.; Xu, Y.; Zhang, H.; Xia, P.; Zhou, W. Tetrahedron: Asymmetry 1999, 10, 2311. doi:10.1016/S0957-4166(99)00239-6 |
| 60. | Liao, L.-X.; Wang, Z.-M.; Zhang, H.-X.; Zhou, W.-S. Tetrahedron: Asymmetry 1999, 10, 3649. doi:10.1016/S0957-4166(99)00382-1 |
| 61. | Haukaas, M. H.; O'Doherty, G. A. Org. Lett. 2001, 3, 401. doi:10.1021/ol006907j |
| 62. | Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2004, 126, 11804. doi:10.1021/ja046185h |
| 63. | Wu, T.-S.; Chan, Y.-Y.; Leu, Y.-L.; Chern, C.-Y.; Chen, C.-F. Phytochemistry 1996, 42, 907. doi:10.1016/0031-9422(95)00962-0 |
| 80. |
Ciufolini, M. A.; Chan, B. K. Heterocycles 2007, 74, 101. doi:10.3987/REV-07-SR(W)4
See for a review. |
| 81. | Ciufolini, M. A.; Roschangar, F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1692. doi:10.1002/anie.199616921 |
| 82. | Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049. doi:10.1016/S0040-4020(97)00365-7 |
| 83. | Ciufolini, M. A.; Roschangar, F. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2000; Vol. 4. |
| 68. | Boger, D. L.; Hong, J. J. Am. Chem. Soc. 1998, 120, 1218. doi:10.1021/ja973007y |
| 69. | Boger, D. L. J. Heterocycl. Chem. 1998, 35, 1003. doi:10.1002/jhet.5570350502 |
| 70. | Bowman, W. R.; Bridge, C. F.; Brookes, P.; Cloonan, M. O.; Leach, D. C. J. Chem. Soc., Perkin Trans. 1 2002, 58. |
| 71. | Kato, I.; Higashimoto, M.; Tamura, O.; Ishibashi, H. J. Org. Chem. 2003, 68, 7983. doi:10.1021/jo030177m |
| 72. | Raolji, G. B.; Garçon, S.; Greene, A. E.; Kanazawa, A. Angew. Chem., Int. Ed. 2003, 42, 5059. doi:10.1002/anie.200352094 |
| 73. | Chavan, S. P.; Sivappa, R. Tetrahedron Lett. 2004, 45, 3941. doi:10.1016/j.tetlet.2004.03.089 |
| 74. | Bowman, W. R.; Cloonan, M. O.; Fletcher, A. J.; Stein, T. Org. Biomol. Chem. 2005, 3, 1460. doi:10.1039/b501509j |
| 75. | Toyota, M.; Komori, C.; Ihara, M. Heterocycles 2000, 52, 591. doi:10.3987/COM-99-S109 |
| 76. | Lin, C. H.; Tsai, M. R.; Wang, Y. S.; Chang, N. C. J. Org. Chem. 2003, 68, 5688. doi:10.1021/jo034042s |
| 77. | Mekouar, K.; Génisson, Y.; Leue, S.; Greene, A. E. J. Org. Chem. 2000, 65, 5212. doi:10.1021/jo0003448 |
| 78. | Toyota, M.; Komori, C.; Ihara, M. J. Org. Chem. 2000, 65, 7110. doi:10.1021/jo000816i |
| 79. | Bowman, W. R.; Bridge, C. F.; Cloonan, M. O.; Leach, D. C. Synlett 2001, 765. doi:10.1055/s-2001-14592 |
| 65. | Kametani, T.; Takeda, H.; Nemoto, H.; Fukumoto, K. J. Chem. Soc., Perkin Trans. 1 1975, 1825. |
| 66. | Pendrak, I.; Barney, S.; Wittrock, R.; Lambert, D. M.; Kingsbury, W. D. J. Org. Chem. 1994, 59, 2623. doi:10.1021/jo00088a057 |
| 67. | Pendrak, I.; Wittrock, R.; Kingsbury, W. D. J. Org. Chem. 1995, 60, 2912. doi:10.1021/jo00114a050 |
© 2011 Turner and Ciufolini; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)