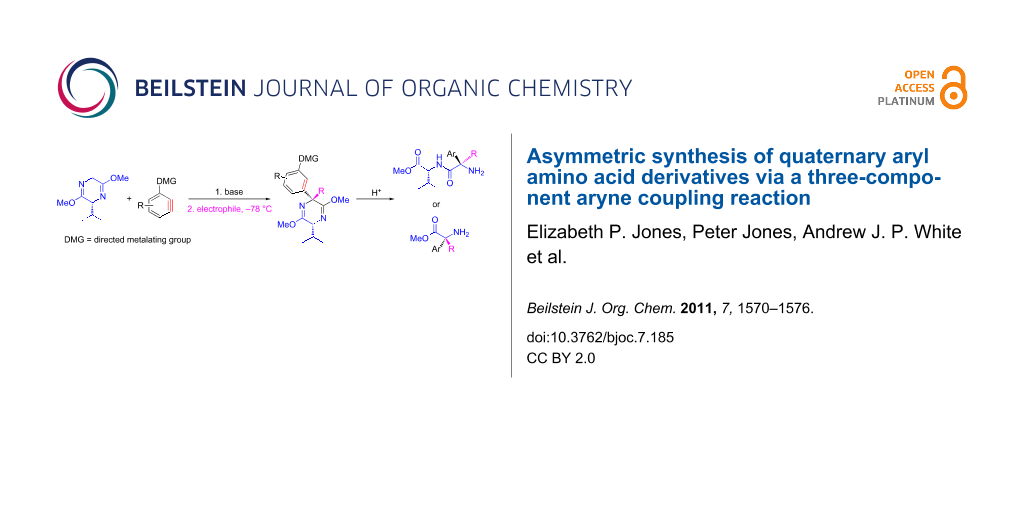
Abstract
A method was developed for the synthesis of α-alkyl, α-aryl-bislactim ethers in good to excellent yields and high diastereoselectivities, consisting of a facile one-pot procedure in which the aryl group is introduced by means of a nucleophilic addition to benzyne and the alkyl group by alkylation of a resultant benzylic anion. Hydrolysis of the sterically less hindered adducts gave the corresponding quaternary amino acids with no racemization, whereas hydrolytic ring opening gave the corresponding valine dipeptides from bulkier bislactims.

Graphical Abstract
Introduction
Arynes are exceptionally versatile reactive intermediates in organic synthesis. Not only are they able to participate in cycloaddition reactions, they also readily undergo addition reactions with nucleophiles, and the resultant aryl anions may be protonated or undergo alternative transformations [1-6]. We are particularly interested in the use of arynes in multicomponent coupling reactions, in which the resultant aryl carbanion following nucleophilic attack is allowed to react with a further electrophile, providing ortho-disubstituted, functionalized aromatic products in a one-pot procedure [7-9]. In particular, we have focused on the addition of carbon-based nucleophiles in such systems (Scheme 1) and have applied this methodology to the syntheses of natural products ent-clavilactone B and dehydroaltenuene B [10,11].
![[1860-5397-7-185-i1]](/bjoc/content/inline/1860-5397-7-185-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 3-Component coupling reactions of arynes. E+ = electrophile.
Scheme 1: 3-Component coupling reactions of arynes. E+ = electrophile.
Recently, we reported the diastereoselective addition of Schöllkopf’s bislactim ether 1 [12] to substituted arynes 2, which, following hydrolysis of the syn-adducts 3, provided α-aryl amino acids 4 with moderate to high enantioselectivities (Scheme 2) [13].
![[1860-5397-7-185-i2]](/bjoc/content/inline/1860-5397-7-185-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Aryne mediated α-arylation of amino acids. DMG = directed metallation group. BHT = 2,6-di-tert-butyl-4-methylphenol.
Scheme 2: Aryne mediated α-arylation of amino acids. DMG = directed metallation group. BHT = 2,6-di-tert-buty...
The arynes were produced by a directed ortho-metallation [14] and an elimination sequence, and after attack of 1 the ensuing carbanion 5 was not aryl as expected but instead benzylic, due to an inter- or intramolecular proton transfer. Kinetic protonation of this anion on the face opposite to the isopropyl group accounted for the observed diastereoselectivity of the reaction (Scheme 3).
![[1860-5397-7-185-i3]](/bjoc/content/inline/1860-5397-7-185-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of α-arylation.
Scheme 3: Proposed mechanism of α-arylation.
To extend this methodology to a multicomponent system, we considered that quenching the key aza-enolate intermediate with an electrophile, rather than a proton source, should provide quaternary Schöllkopf adducts 6 (Scheme 4).
![[1860-5397-7-185-i4]](/bjoc/content/inline/1860-5397-7-185-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed extension of the methodology to synthesize quaternary adducts.
Scheme 4: Proposed extension of the methodology to synthesize quaternary adducts.
The potential utility of chiral α-quaternary α-aryl amino acids in medicinal chemistry is diverse. Incorporation of such units into peptidomimetics, not only affects lipophilicity, but also the secondary structure and hence the conformational rigidity, which can increase the resistance to enzymatic degradation [15-19]. Such units are also found in biologically interesting natural products, such as the antibacterial fumimycin [20].
Herein, we describe our endeavours towards the synthesis of such valuable compounds by utilizing the multicomponent aryne reaction.
Results and Discussion
Formation of quaternary Schöllkopf adducts
The same procedure was carried out as for the α-arylation reactions: The benzyne precursor 2a and Schöllkopf’s bislactim ether 1 were allowed to react with 2.5 equivalents of sec-butyllithium at –95 °C to carry out the required ortho-lithiation for benzyne formation from 2a and the deprotonation of bislactim 1. During warming to room temperature, fragmentation to dimethoxybenzyne occurred and nucleophilic attack of the Schöllkopf auxiliary took place. Addition of four equivalents of iodomethane at this temperature and stirring for 1 h gave the desired quaternary species 6a in 88% yield with a dr of 85:15. Lowering the quench temperature to –78 °C increased both the yield and the dr to 92% and 96:4, respectively (Scheme 5).
![[1860-5397-7-185-i5]](/bjoc/content/inline/1860-5397-7-185-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Formation of α-methyl, α-aryl Schöllkopf adduct.
Scheme 5: Formation of α-methyl, α-aryl Schöllkopf adduct.
The major diastereoisomer was confirmed to have the isopropyl and aryl moieties syn, by 1H NMR NOESY analysis; a clear NOESY correlation was observed between the methyl group and the proton at C-3 (Figure 1).
![[1860-5397-7-185-1]](/bjoc/content/figures/1860-5397-7-185-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: NOESY correlation observed for 6a.
Figure 1: NOESY correlation observed for 6a.
The scope of the reaction was then examined to determine which electrophiles could be introduced at C-6 (Table 1).
Table 1: Formation of quaternary Schöllkopf adducts employing a range of electrophiles.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-185-i11.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | Electrophile | R | Time (h)b | Product | Yield (%)c | drd |
|---|---|---|---|---|---|---|
| 1 | MeI | Me | 1 | 6a | 92 | 96:4 |
| 2 | BnBr | Bn | 6 | 6b | 88 | >98:2 |
| 3 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-185-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-185-i13.svg?max-width=637&scale=1.0)
|
1 | 6c | 85 | >98:2 |
| 4 | MOMCl |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-185-i14.svg?max-width=637&scale=1.0)
|
1 | 6d | 53 | 95:5 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-185-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-185-i16.svg?max-width=637&scale=1.0)
|
1 | 6e | 80 | >98:2 |
| 6 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-185-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-185-i18.svg?max-width=637&scale=1.0)
|
3 | 6f | 51 | >98:2 |
| 7 | AcCl | COMe | 6 | 6g | 59 | 89:11 |
| 8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-185-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-185-i20.svg?max-width=637&scale=1.0)
|
5 | 6h | 71 | >98:2 |
| 9 | PhCHO |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-185-i21.svg?max-width=637&scale=1.0)
|
12 | 6ie | 69 | >98:2f |
| 10 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-185-i22.svg?max-width=637&scale=1.0)
BF3·OEt2 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-185-i23.svg?max-width=637&scale=1.0)
|
1 | 6j | 50 | >98:2 |
aReactions were carried out on a 1 mmol scale in THF (0.2 M) with 1.5 equiv of halide 2a and 2.75 equiv s-BuLi at –95 °C, followed by warming to room temperature over 18 h, recooling to –78 °C, and the addition of 4 equiv of electrophile. bReactions were monitored by GC–MS. cIsolated yield. dDetermined by 1H NMR integration. e6i was isolated as its benzoyl ester, due to retro-aldol reactions occurring when the free alcohol was subjected to flash-column chromatography. fThe adduct 6i was obtained as a mixture of epimeric benzylic alcohols (1:1).
Alkylations employing benzyl bromide, allyl bromide and propargyl bromide (Table 1, entries 2, 3 and 5) proceeded smoothly, furnishing the desired products 6b, 6c and 6e in excellent yields and high diastereoselectivity. Interestingly, adduct 6b displays a large upfield shift (around 2 ppm) at the C-3 proton compared to other analogues. This indicates that the product has adopted an “aryl-inside” conformation in which the C-3 proton is situated in the shielding cone of the aromatic ring [21]. A single-crystal X-ray crystallographic analysis confirmed the presence of this conformation, at least in the crystal lattice (Figure 2).
![[1860-5397-7-185-2]](/bjoc/content/figures/1860-5397-7-185-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Further alkylations with methyl chloromethyl ether, tert-butyl bromoacetate and 2-(bromomethyl)-6-methylpyridine (Table 1, entries 4, 6 and 8) provided adducts 6d, 6f and 6h respectively, also with high diastereoselectivity, but in more modest yields. Acylation with acetyl chloride (Table 1, entry 7) afforded ketone 6g in 59% yield, with a lower dr of 89:11, most likely due to the less sterically hindered nature of this electrophile. Replacing acetyl chloride with its corresponding Weinreb amide gave none of the required product. An aldol type reaction with benzaldehyde (Table 1, entry 9) afforded alcohol 6i in good yield, with complete diastereoselectivity at C-6, but no selectivity at the alcohol stereocenter (1:1). Examination of the two possible Zimmerman–Traxler type transition states for such reactions (Figure 3), as proposed by Schöllkopf [22], reveals that TS≠ A, with the phenyl group axial, is consistent with there being an unfavourable 1,3-diaxial interaction with the methoxy group. However, there is an equally unfavourable interaction in TS≠ B where the phenyl group occupies the equatorial position, as there is now a 1,2-gauche interaction with the bulky di-methoxyaryl group adjacent to it. We consider that neither of these transition states is lower in energy, which would be consistent with the lack of diastereoselectivity.
![[1860-5397-7-185-3]](/bjoc/content/figures/1860-5397-7-185-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Transition state analysis to explain the lack of diastereoselectivity at C-2.
Figure 3: Transition state analysis to explain the lack of diastereoselectivity at C-2.
Finally, the addition of (R)-propylene oxide as the electrophile in the presence of boron trifluoride etherate (Table 1, entry 10) gave alcohol 6j. When (S)-propylene oxide was used instead there was no reaction, and indeed Schöllkopf noted significant levels of kinetic resolution when such reactions were performed with racemic oxiranes [23].
In our previous publication, we demonstrated the scope of the methodology over a range of ortho-lithiation benzyne precursors. To establish that any one of these precursors could be used to form quaternary adducts, we subjected the benzylation conditions to precursor 2b and the allylation conditions to precursor 2c, affording adducts 7b and 7c, respectively, again in good yields and dr’s and with complete regioselectivity for these asymmetric benzynes (Scheme 6).
![[1860-5397-7-185-i6]](/bjoc/content/inline/1860-5397-7-185-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of quaternary adducts.
Scheme 6: Formation of quaternary adducts.
Hydrolysis of quaternary adducts
The next task to be undertaken was the hydrolysis of the sterically restricted quaternary bislactim ethers. A selection of the relatively less bulky products, 6a, 6d and 6e, was subjected to standard, mild hydrolysis conditions, employing 0.5 M HCl in THF at room temperature [12], affording the desired quaternary methyl esters as their corresponding dipeptides, 8a, 8d and 8e, in good yields (Scheme 7). The less shielded imidate group underwent complete hydrolysis, but the imidate functional group α to the quaternary center experienced incomplete cleavage to the peptide bond, a trend which is common in the literature for quaternary adducts [21,24-26]. Subjecting analogue 7c, with just one methoxy group on the aromatic ring, to the same conditions yielded the dipeptide 9 and the desired methyl ester 10 in a 1:1 ratio (Scheme 7).
![[1860-5397-7-185-i7]](/bjoc/content/inline/1860-5397-7-185-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Hydrolysis of quaternary adducts.
Scheme 7: Hydrolysis of quaternary adducts.
Screening of various acidic hydrolysis reaction conditions in these systems led to the observation that when either 6a or 7c was stirred in neat 6 M sulfuric acid for three days, the required constituent amino acid 11 or 10 was isolated in reasonable yields (Scheme 8). Chiral HPLC analysis confirmed the expected high enantioselectivity of these esters.
![[1860-5397-7-185-i8]](/bjoc/content/inline/1860-5397-7-185-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
These results are promising, as they demonstrate that if the aryl group is derived from a less sterically hindered benzyne precursor, hydrolysis is more facile, as was observed by Lee [27] for unsubstituted aryl, quaternary bislactim ethers. Using high concentrations of acids with more-nucleophilic conjugate bases resulted in the predominate formation of diketopiperazines, and low concentrations of all of the acids tested gave the dipeptide as the major product for all analogues with two methoxy groups (6a–6i).
The only example where the use of low concentrations of acid coupled with a dimethoxy aryl group resulted in the principal formation of the desired ester was in the case of alcohol 6j. Mild acidic hydrolysis with 0.5 M HCl gave lactone 12 in 80% yield and 96% ee (Scheme 9). The ability of the alcohol to undergo lactonization under the reaction conditions clearly aided the hydrolysis of this more sterically hindered imidate.
![[1860-5397-7-185-i9]](/bjoc/content/inline/1860-5397-7-185-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
We assumed that ketone 6g would also undergo hydrolysis due to its nonbulky nature. However, upon exposure to 0.5 M HCl, epimerization of the isopropyl group at C-3 occurs in near quantitative yield, giving adduct 13 (Scheme 10). The electron-withdrawing effect of the carbonyl moiety must increase the acidity of the C-3 proton to such an extent that the acidic media simply epimerizes at this center to afford the more thermodynamically stable anti-adduct.
![[1860-5397-7-185-i10]](/bjoc/content/inline/1860-5397-7-185-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
The extension of the aryne α-arylation methodology allowed for the synthesis of quaternary aryl bislactim ethers in good to excellent yields and high diastereoselectivity by means of a three-component coupling reaction in which the reaction of an electrophile with the intermediate benzylic anion gave a range of C–alkyl and hydroxyalkyl derivatives. Hydrolysis of these quaternary adducts gave the constituent amino acid methyl esters in high enantioselectivity for less substituted compounds. Adducts with two methyl ethers on the aryl unit underwent hydrolysis to the corresponding valine dipeptides.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and analytical data for compounds 6a–j, 7b–c, 8a, 8d, 8e and 9–13. | ||
| Format: PDF | Size: 358.2 KB | Download |
| Supporting Information File 2: NMR spectral data for compounds 6a–j and 7b–c. | ||
| Format: PDF | Size: 461.2 KB | Download |
References
-
Pellissier, H.; Santelli, M. Tetrahedron 2003, 59, 701–730. doi:10.1016/S0040-4020(02)01563-6
Return to citation in text: [1] -
Peña, D.; Pérez, D.; Guitián, E. Angew. Chem., Int. Ed. 2006, 45, 3579–3581. doi:10.1002/anie.200600291
Return to citation in text: [1] -
Sanz, R. Org. Prep. Proced. Int. 2008, 40, 215–291. doi:10.1080/00304940809458089
Return to citation in text: [1] -
Liu, Z.; Larock, R. C. Org. Lett. 2003, 5, 4673–4675. doi:10.1021/ol0358612
Return to citation in text: [1] -
Rémond, E.; Tessier, A.; Leroux, F. R.; Bayardon, J.; Jugé, S. Org. Lett. 2010, 12, 1568–1571. doi:10.1021/ol100304c
Return to citation in text: [1] -
Liu, Z.; Larock, R. C. Org. Lett. 2004, 6, 99–102. doi:10.1021/ol0361406
Return to citation in text: [1] -
Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. Angew. Chem., Int. Ed. 2004, 43, 3935–3938. doi:10.1002/anie.200460009
See this reference and [8,9] for examples of 3-component coupling reactions of arynes.
Return to citation in text: [1] -
Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. J. Am. Chem. Soc. 2006, 128, 11040–11041. doi:10.1021/ja064157o
Return to citation in text: [1] [2] -
Yoshida, H.; Morishita, T.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2007, 9, 3367–3370. doi:10.1021/ol071347a
Return to citation in text: [1] [2] -
Larrosa, I.; Da Silva, M. I.; Gómez, P. M.; Hannen, P.; Ko, E.; Lenger, S. R.; Linke, S. R.; White, A. J. P.; Wilton, D.; Barrett, A. G. M. J. Am. Chem. Soc. 2006, 128, 14042–14043. doi:10.1021/ja0662671
Return to citation in text: [1] -
Soorukram, D.; Qu, T.; Barrett, A. G. M. Org. Lett. 2008, 10, 3833–3835. doi:10.1021/ol8015435
Return to citation in text: [1] -
Schöllkopf, U. Tetrahedron 1983, 39, 2085–2091. doi:10.1016/S0040-4020(01)91926-X
Return to citation in text: [1] [2] -
Jones, E. P.; Jones, P.; Barrett, A. G. M. Org. Lett. 2011, 13, 1012–1015. doi:10.1021/ol1030469
Return to citation in text: [1] -
Kaelin, D. E., Jr.; Lopez, O. D.; Martin, S. F. J. Am. Chem. Soc. 2001, 123, 6937–6938. doi:10.1021/ja0108640
Return to citation in text: [1] -
Tanaka, M. Chem. Pharm. Bull. 2007, 55, 349–358. doi:10.1248/cpb.55.349
Return to citation in text: [1] -
Toniolo, C.; Formaggio, F.; Kaptein, B.; Broxterman, Q. B. Synlett 2006, 1295–1310. doi:10.1055/s-2006-941573
Return to citation in text: [1] -
Karle, I. L.; Kaul, R.; Rao, R. B.; Raghothama, S.; Balaram, P. J. Am. Chem. Soc. 1997, 119, 12048–12054. doi:10.1021/ja970596z
Return to citation in text: [1] -
Crisma, M.; Valle, G.; Bonora, G. M.; Toniolo, C.; Lelj, F.; Barone, V.; Fraternall, F.; Hardy, P. M.; Maia, H. L. S. Biopolymers 1991, 31, 637–641. doi:10.1002/bip.360310608
Return to citation in text: [1] -
Gante, J. Angew. Chem., Int. Ed. Engl. 1994, 33, 1699–1720. doi:10.1002/anie.199416991
Return to citation in text: [1] -
Kwon, Y.-J.; Sohn, M.-J.; Zheng, C.-J.; Kim, W.-G. Org. Lett. 2007, 9, 2449–2451. doi:10.1021/ol0703231
Return to citation in text: [1] -
Schöllkopf, U.; Busse, U.; Kilger, R.; Lehr, P. Synthesis 1984, 271–274.
Return to citation in text: [1] [2] -
Schöllkopf, U. Pure Appl. Chem. 1983, 55, 1799–1806. doi:10.1351/pac198355111799
Return to citation in text: [1] -
Gull, R.; Schöllkopf, U. Synthesis 1985, 1052–1055. doi:10.1055/s-1985-31425
Return to citation in text: [1] -
Krikstolaityte, S.; Sackus, A.; Romming, C.; Undheim, K. Chemija 2009, 20, 124–127.
Return to citation in text: [1] -
Andrei, M.; Römming, C.; Undheim, K. Tetrahedron: Asymmetry 2004, 15, 2711–2717. doi:10.1016/j.tetasy.2004.07.030
Return to citation in text: [1] -
Hammer, K.; Undheim, K. Tetrahedron: Asymmetry 1998, 9, 2359–2368. doi:10.1016/S0957-4166(98)00248-1
Return to citation in text: [1] -
Lee, S.-H.; Lee, E.-K. Bull. Korean Chem. Soc. 2001, 22, 551–552.
Return to citation in text: [1]
| 1. | Pellissier, H.; Santelli, M. Tetrahedron 2003, 59, 701–730. doi:10.1016/S0040-4020(02)01563-6 |
| 2. | Peña, D.; Pérez, D.; Guitián, E. Angew. Chem., Int. Ed. 2006, 45, 3579–3581. doi:10.1002/anie.200600291 |
| 3. | Sanz, R. Org. Prep. Proced. Int. 2008, 40, 215–291. doi:10.1080/00304940809458089 |
| 4. | Liu, Z.; Larock, R. C. Org. Lett. 2003, 5, 4673–4675. doi:10.1021/ol0358612 |
| 5. | Rémond, E.; Tessier, A.; Leroux, F. R.; Bayardon, J.; Jugé, S. Org. Lett. 2010, 12, 1568–1571. doi:10.1021/ol100304c |
| 6. | Liu, Z.; Larock, R. C. Org. Lett. 2004, 6, 99–102. doi:10.1021/ol0361406 |
| 13. | Jones, E. P.; Jones, P.; Barrett, A. G. M. Org. Lett. 2011, 13, 1012–1015. doi:10.1021/ol1030469 |
| 8. | Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. J. Am. Chem. Soc. 2006, 128, 11040–11041. doi:10.1021/ja064157o |
| 9. | Yoshida, H.; Morishita, T.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2007, 9, 3367–3370. doi:10.1021/ol071347a |
| 12. | Schöllkopf, U. Tetrahedron 1983, 39, 2085–2091. doi:10.1016/S0040-4020(01)91926-X |
| 10. | Larrosa, I.; Da Silva, M. I.; Gómez, P. M.; Hannen, P.; Ko, E.; Lenger, S. R.; Linke, S. R.; White, A. J. P.; Wilton, D.; Barrett, A. G. M. J. Am. Chem. Soc. 2006, 128, 14042–14043. doi:10.1021/ja0662671 |
| 11. | Soorukram, D.; Qu, T.; Barrett, A. G. M. Org. Lett. 2008, 10, 3833–3835. doi:10.1021/ol8015435 |
| 21. | Schöllkopf, U.; Busse, U.; Kilger, R.; Lehr, P. Synthesis 1984, 271–274. |
| 24. | Krikstolaityte, S.; Sackus, A.; Romming, C.; Undheim, K. Chemija 2009, 20, 124–127. |
| 25. | Andrei, M.; Römming, C.; Undheim, K. Tetrahedron: Asymmetry 2004, 15, 2711–2717. doi:10.1016/j.tetasy.2004.07.030 |
| 26. | Hammer, K.; Undheim, K. Tetrahedron: Asymmetry 1998, 9, 2359–2368. doi:10.1016/S0957-4166(98)00248-1 |
| 7. |
Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. Angew. Chem., Int. Ed. 2004, 43, 3935–3938. doi:10.1002/anie.200460009
See this reference and [8,9] for examples of 3-component coupling reactions of arynes. |
| 8. | Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. J. Am. Chem. Soc. 2006, 128, 11040–11041. doi:10.1021/ja064157o |
| 9. | Yoshida, H.; Morishita, T.; Fukushima, H.; Ohshita, J.; Kunai, A. Org. Lett. 2007, 9, 3367–3370. doi:10.1021/ol071347a |
| 20. | Kwon, Y.-J.; Sohn, M.-J.; Zheng, C.-J.; Kim, W.-G. Org. Lett. 2007, 9, 2449–2451. doi:10.1021/ol0703231 |
| 12. | Schöllkopf, U. Tetrahedron 1983, 39, 2085–2091. doi:10.1016/S0040-4020(01)91926-X |
| 15. | Tanaka, M. Chem. Pharm. Bull. 2007, 55, 349–358. doi:10.1248/cpb.55.349 |
| 16. | Toniolo, C.; Formaggio, F.; Kaptein, B.; Broxterman, Q. B. Synlett 2006, 1295–1310. doi:10.1055/s-2006-941573 |
| 17. | Karle, I. L.; Kaul, R.; Rao, R. B.; Raghothama, S.; Balaram, P. J. Am. Chem. Soc. 1997, 119, 12048–12054. doi:10.1021/ja970596z |
| 18. | Crisma, M.; Valle, G.; Bonora, G. M.; Toniolo, C.; Lelj, F.; Barone, V.; Fraternall, F.; Hardy, P. M.; Maia, H. L. S. Biopolymers 1991, 31, 637–641. doi:10.1002/bip.360310608 |
| 19. | Gante, J. Angew. Chem., Int. Ed. Engl. 1994, 33, 1699–1720. doi:10.1002/anie.199416991 |
| 14. | Kaelin, D. E., Jr.; Lopez, O. D.; Martin, S. F. J. Am. Chem. Soc. 2001, 123, 6937–6938. doi:10.1021/ja0108640 |
| 22. | Schöllkopf, U. Pure Appl. Chem. 1983, 55, 1799–1806. doi:10.1351/pac198355111799 |
© 2011 Jones et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)