Abstract
The synthesis of multifunctional indenes with at least two different functional groups has not yet been extensively explored. Among the plausible synthetic routes to 3,5-disubstituted indenes bearing two different functional groups, such as the [3-(aminoethyl)inden-5-yl)]amines, a reasonable pathway involves the (5-nitro-3-indenyl)acetamides as key intermediates. Although several multistep synthetic approaches can be applied to obtain these advanced intermediates, we describe herein their preparation by an aldol-type reaction between 5-nitroindan-1-ones and the lithium salt of N,N-disubstituted acetamides, followed immediately by dehydration with acid. This classical condensation process, which is neither simple nor trivial despite its apparent directness, permits an efficient entry to a variety of indene-based molecular modules, which could be adapted to a range of functionalized indanones.

Graphical Abstract
Introduction
Compounds with an indene core are of great interest as precursors of metallocene complexes for catalytic polymerization processes, as well as being present in N-heterocyclic carbene ligands and functional materials [1-9]. In addition, indene-based structures are a source of bioactive compounds in drug discovery and development [10-18]. The routes to access multiply substituted indenes with at least two different functional groups (FGs) are generally complex, and the synthetic approaches leading to these compounds lag behind those for heteroaromatic systems, e.g., indoles, being limited as it is by the scarce knowledge of indene chemistry in comparison with heterocyclic chemistry [3,17-22]. Moreover, chemical transformation by indenes, which could permit rapid access to different substitution patterns, is rather cumbersome owing to the complexity of the indene chemistry, especially of those processes that could be modulated by the aromatic character of a resulting indenyl anion species. Indeed, indene is an unusually acidic nonaromatic carbocyclic system and, although structurally different, indene and indole show similar pKa values of 20.1 and 21.0 in dimethylsulfoxide solution, respectively [23,24].
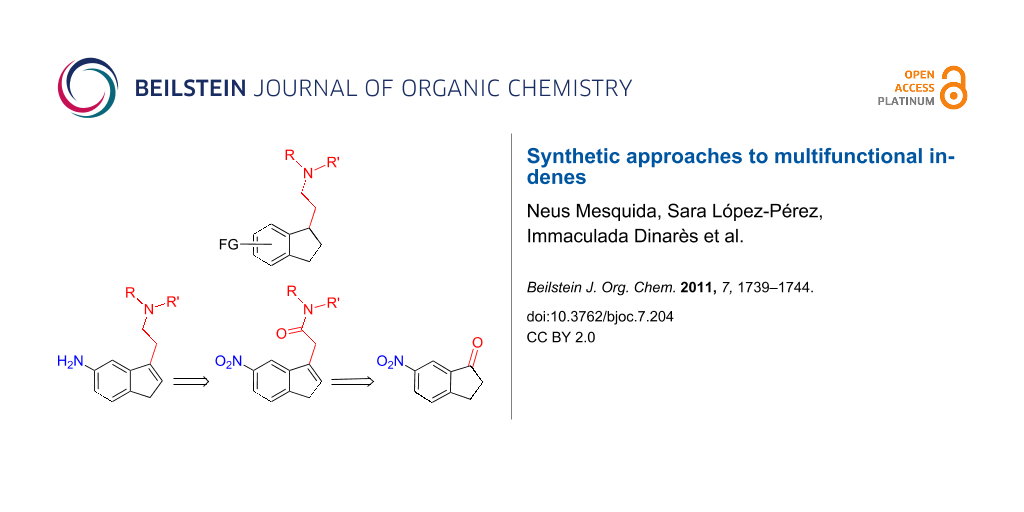
During the course of our studies on indene-based ligands of general type 1 with biological effects on the central nervous system, we found that different stepwise synthetic routes could be applied to inden-5-amines 2 bearing a disubstituted N,N-aminoethyl side arm at the indene 3-position [17,18]. After analyzing reasonable synthetic routes to the [3-(aminoethyl)inden-5-yl)]amine intermediates 2 starting from substituted indan-1-one 5, route A was chosen as the most suitable way of functionalizing the 5-position of the indene, e.g., by changing the aryl(heteroaryl) structural motif of a sulfonamide moiety (Scheme 1). This route proceeded in three steps, and allowed the representative (3-indenyl)acetic acids 4 to be conveniently prepared from nitroindanones 5 and the lithium salt of ethyl acetate [18].
![[1860-5397-7-204-i1]](/bjoc/content/inline/1860-5397-7-204-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic pathways to 3,5-disubstituted indenes bearing two functional groups: Indenylsulfonamides 1.
Scheme 1: Retrosynthetic pathways to 3,5-disubstituted indenes bearing two functional groups: Indenylsulfonam...
The present work deals with the alternative synthetic route B, which relies on the direct formation of the advanced (3-indenyl)acetamides 3 from nitroindanone 5 and shortens the synthetic sequence to two steps. The aldol-type reaction of indanone 5 with the lithium salts of different N,N-disubstituted acetamides, followed immediately by dehydration afforded the (3-indenyl)acetamides 3. The success of this route, however, depends on two main factors: (i) The nature of the lithiated base used to deprotonate acetamides in order to form lithium enolates, which needs to be considered when adjusting the reaction conditions [25]; and (ii) the chemical response of indanoles towards dehydration [6,18] to give the desired endo-olefin 3, which should be favored with the presence of a methyl group at the indene 2-position.
Results and Discussion
We recently reported that the reaction of 2-methylindanone 6 with the lithium salt of ethyl acetate (generated in situ by the reaction of lithium hexamethyldisilazide with ethyl acetate), immediately followed by dehydration and hydrolysis/isomerization, afforded the acetic acid 7 with an improved yield of 74%. Compound 7 was then conveniently transformed to the corresponding acetamide 8. The reduction of the amide group with AlH3·NMe2Et was the critical step for the preparation of the key inden-5-amine 9 due to the particularly troublesome isolation process [18].
Thus, a different synthetic option was examined in which the (3-indenyl)acetic acid 7 was reduced to the corresponding alcohol 10 and transformed to tosylate 11. However, an attempt to convert the (3-indenyl)ethylsulfonate 11 into the advanced inden-5-amine 9 was ineffective, resulting in the formation of the spiro indene 12 instead, and hence this route was not studied further (Scheme 2 and Supporting Information File 1). It should be mention that the propensity of several 3-substituted indenes, appropriately fitted with leaving groups, to undergo spirocyclization has been previously reported [26,27].
![[1860-5397-7-204-i2]](/bjoc/content/inline/1860-5397-7-204-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (i) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O, 60 °C [18]; (ii) (a) SOCl2, CH2Cl2, reflux, (b) Me2NH, rt [18]; (iii) (a) AlH3·NMe2Et, THF, 0 °C, (b) Zn, AcOH, rt [18]; (iv) AlH3·NMe2Et, THF, 0 °C; (v) TsCl, Py, CH2Cl2, rt; (vi) Me2NH, DMF, rt; (vii) (a) N,N-dimethylacetamide, LDA, THF, −78 °C, (b) TFA, CH2Cl2, rt, 2 h.
Scheme 2: Reagents and conditions: (i) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O, 60 °C [18]; (ii) (a) SOCl2,...
To shorten the multistep route A, an alternative preparation of the acetamide 8 was examined, based on an aldol-type reaction between nitroindanone 6 and the lithium salt of N,N-dimethylacetamide. After various reaction conditions and lithiated bases were examined, the condensation of 6 with the α-lithio-N,N-dimethylacetamide (prepared in turn from N,N-dimethylacetamide and an excess of lithium diisopropylamide), immediately followed by dehydration with trifluoroacetic acid, afforded indenylacetamide 8 in 70% yield, which was 14% higher than the previously reported result obtained with the (3-indenyl)acetic acid 7 [18]. Upon application of the same experimental conditions to indanone 13, the yield of indenylacetamide 15 was improved from 37% [18] to 65% (Scheme 2 and Supporting Information File 1).
We next studied the utility of this aldol-type reaction protocol by using other disubstituted acetamides, such as N-acetylpyrrolidine and N-acetylpiperidine, as shown in Scheme 3. When the reaction conditions used to prepare N,N-dimethyl-(3-indenyl)acetamide 8 were applied, 2-methylindanone 6 failed to give the (3-indenyl)acetamide 16, and did not progress beyond the corresponding indanol (Supporting Information File 1). When the dehydration process was prolonged to 17 h at room temperature, conversion of indanone 6 to the desired acetamide 16 proceeded in 74% yield, which was 14% higher than that previously reported for a two-step procedure with the (3-indenyl)acetic acid 7 [18]. Following this modified procedure, the reaction between indanones 6 or 13 and the lithium salt of N-acetylpiperidine or N-acetylpyrrolidine afforded indenylacetamides 17–19 in good yield (≥67%).
![[1860-5397-7-204-i3]](/bjoc/content/inline/1860-5397-7-204-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (i) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O, 60 °C [18]; (ii) (a) SOCl2, CH2Cl2, reflux, (b) pyrrolidine or piperidine, rt [7]; (iii) (a) N-acetylpyrrolidine or N-acetylpiperidine, LDA, −78 °C; (b) TFA, CH2Cl2, rt, 17 h.
Scheme 3: Reagents and conditions: (i) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O, 60 °C [18]; (ii) (a) SOCl2,...
Finally, to broaden the scope of this aldol-type reaction, we examined the synthesis of (1,1-dimethyl-3-indenyl)acetamides. Following a standard nitration protocol applied to different indanones [17], the 3,3-dimethylindanone 20 was efficiently transformed into the corresponding 6-nitroindan-1-one 21 in 97% yield and no traces of the isomer 4-nitroindanone were detected, probably due to the presence of the dimethyl moiety at the indanone 3-position in 20. This was a marked improvement on a previously reported 62% yield of nitroindanone 21 employing a different nitration method [28]. In our recently reported preliminary experiments, the conversion of 21 to the corresponding (3-indenyl)acetic acid 22 resulted in a fairly low yield of 27%, and the transformation to N,N-dimethyl-(3-indenyl)acetamide 23 was not carried out [18]. However, after the synthetic approach was changed from route A to route B, the indenylacetamide 23 was prepared by aldol-type condensation between indanone 21 and the lithium salt of N,N-dimethylacetamide, in 57% yield (Scheme 4).
![[1860-5397-7-204-i4]](/bjoc/content/inline/1860-5397-7-204-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: (i) KNO3, H2SO4, −5 °C; (ii) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O, 60 °C, 7 h [18]; (iii) (a) N,N-dimethylacetamide, LDA, THF, −78 °C, (b) TFA, CH2Cl2, rt, 17 h.
Scheme 4: Reagents and conditions: (i) KNO3, H2SO4, −5 °C; (ii) (a) EtOAc, LHMDS, THF, −78 °C, (b) H2SO4, H2O...
We thus successfully optimized the synthesis of (3-indenyl)acetamides 3, obtaining higher yields with a shorter procedure.
The crude reaction products of the aldol-type reaction between nitroindanones and α-lithioacetamides often contained minor amounts of exo-olefins. Pure indenylacetamides were isolated after careful column chromatography, and sometimes a high-throughput flash purification system was necessary.
The structures of the new compounds were confirmed by spectroscopic methods, and their 1H NMR and 13C NMR chemical shifts and physical data are gathered in the experimental description (Supporting Information File 1). The constitution of the acetamides 17, 19 and 23 was determined by 1D NOESY experiments in CDCl3 at 500 MHz. Thus, for each compound, irradiation at the H-2 proton of the indene core revealed a NOE for the methylene protons H-1 and H-a. For compound 23, an additional NOE was observed at the methyl protons of the acetamide group (Figure 1).
![[1860-5397-7-204-1]](/bjoc/content/figures/1860-5397-7-204-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Key NMR responses for compounds 17, 19 and 23: 1D NOESY experiments.
Figure 1: Key NMR responses for compounds 17, 19 and 23: 1D NOESY experiments.
Conclusion
Among different synthetic routes that could lead to disubstituted indenes 1 bearing two functional groups, such as the indenamines 2, a feasible path explored here required the use of (5-nitro-3-indenyl)acetamides 3, prepared either from (5-nitro-3-indenyl)acetic acid (4) or 5-nitroindan-1-one (5). The best option to prepare the advanced indenylacetamides 3 was based on an aldol-type reaction between nitroindanone 5 and the lithium salt of N,N-disubstituted acetamides, followed immediately by dehydration with acid. Although apparently direct, the classical aldol-type condensation process is neither simple nor rudimentary when applied to members of the indene family. Particularly significant aspects of the reported transformation are (i) the nature of the lithiated base used to form the lithium enolate, and (ii) the dehydration process to obtain the desired endo-olefin 3. The optimal experimental results were achieved with lithium diisopropylamide as the base and by employing a long dehydration reaction time, which provided the targeted indenylacetamides 3 in good yield. This synthetic protocol can be exploited for the elaboration of a variety of indene-based molecular modules with applications in fields as diverse as bioactive compounds, ligand precursors for metallocene catalyst systems, and functional materials.
Supporting Information
| Supporting Information File 1: Assays related to the preparation of 8, 10, 11 and 16, experimental details, characterization data and copies of NMR and ESI-HRMS spectra of all new compounds. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
This research was supported by Vicerrectorat de Recerca, Universitat de Barcelona and by the D. G. I. (MICINN) Projects CTQ2006-1182/BQU and CTQ2010-15251/BQU. Thanks are also due to the AGAUR (Generalitat de Catalunya), Grup de Recerca Consolidat 2009SGR562. S. L.-P. is grateful to the Agaur for a 2008-F.I. fellowship.
References
-
Chirik, P. J. Organometallics 2010, 29, 1500–1517. doi:10.1021/om100016p
Return to citation in text: [1] -
Zhang, C.; Luo, F.; Cheng, B.; Li, B.; Song, H.; Xu, S.; Wang, B. Dalton Trans. 2009, 7230–7235. doi:10.1039/b906933j
Return to citation in text: [1] -
Guo, S.; Liu, Y. Org. Biomol. Chem. 2008, 6, 2064–2070. doi:10.1039/b804888f
And references therein.
Return to citation in text: [1] [2] -
Guan, Z.-H.; Ren, Z.-H.; Zhao, L.-B.; Liang, Y.-M. Org. Biomol. Chem. 2008, 6, 1040–1045. doi:10.1039/b719957k
Return to citation in text: [1] -
Enders, M.; Baker, R. W. Curr. Org. Chem. 2006, 10, 937–953. doi:10.2174/138527206777435508
Return to citation in text: [1] -
Silver, S.; Leppänen, A.-S.; Sjöholm, R.; Penninkangas, A.; Leino, R. Eur. J. Org. Chem. 2005, 1058–1081. doi:10.1002/ejoc.200400789
Return to citation in text: [1] [2] -
Leino, R.; Lehmus, P.; Lehtonen, A. Eur. J. Inorg. Chem. 2004, 3201–3222. doi:10.1002/ejic.200400214
Return to citation in text: [1] [2] -
Zargarian, D. Coord. Chem. Rev. 2002, 233–234, 157–176. doi:10.1016/S0010-8545(02)00201-1
Return to citation in text: [1] -
Hummel, S.; Kirsch, S. F. Beilstein J. Org. Chem. 2011, 7, 847–859. doi:10.3762/bjoc.7.97
Return to citation in text: [1] -
Waldmann, H.; Karaguni, I.-M.; Carpintero, M.; Gourzoulidou, E.; Herrmann, C.; Brockmann, C.; Oschkinat, H.; Müller, O. Angew. Chem., Int. Ed. 2004, 43, 454–458. doi:10.1002/anie.200353089
Return to citation in text: [1] -
Karaguni, I.-M.; Glüsenkamp, K.-H.; Langerak, A.; Geisen, C.; Ullrich, V.; Winde, G.; Möröy, T.; Müller, O. Bioorg. Med. Chem. Lett. 2002, 12, 709–713. doi:10.1016/S0960-894X(01)00839-3
Return to citation in text: [1] -
Li, B.-F.; Moree, W. J.; Yu, J.; Coon, T.; Zamani-Kord, S.; Malany, S.; Jalali, K.; Wen, J.; Wang, H.; Yang, C.; Hoare, S. R. J.; Petroski, R. E.; Madan, A.; Crowe, P. D.; Beaton, G. Bioorg. Med. Chem. Lett. 2010, 20, 2629–2633. doi:10.1016/j.bmcl.2010.02.055
Return to citation in text: [1] -
Moree, W. J.; Li, B.-F.; Jovic, F.; Coon, T.; Yu, J.; Gross, R. S.; Tucci, F.; Marinkovic, D.; Zamani-Kord, S.; Malany, S.; Bradbury, M. J.; Hernandez, L. M.; O’Brien, Z.; Wen, J.; Wang, H.; Hoare, S. R. J.; Petroski, R. E.; Sacaan, A.; Madan, A.; Crowe, P. D.; Beaton, G. J. Med. Chem. 2009, 52, 5307–5310. doi:10.1021/jm900933k
Return to citation in text: [1] -
Hodgson, D. M.; Winning, L. H. Beilstein J. Org. Chem. 2008, 4, No. 38. doi:10.3762/bjoc.4.38
Return to citation in text: [1] -
Huffman, J. W.; Padgett, L. W. Curr. Med. Chem. 2005, 12, 1395–1411. doi:10.2174/0929867054020864
Return to citation in text: [1] -
Böhme, T. M.; Keim, C.; Kreutzmann, K.; Linder, M.; Dingermann, T.; Dannhardt, G.; Mutschler, E.; Lambrecht, G. J. Med. Chem. 2003, 46, 856–867. doi:10.1021/jm020895l
Return to citation in text: [1] -
Alcalde, E.; Mesquida, N.; Frigola, J.; López-Pérez, S.; Mercè, R. Org. Biomol. Chem. 2008, 6, 3795–3810. doi:10.1039/b808641a
And references cited therein.
Return to citation in text: [1] [2] [3] [4] -
Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] -
Wang, J.; Zhang, L.; Jing, Y.; Huang, W.; Zhou, X. Tetrahedron Lett. 2009, 50, 4978–4982. doi:10.1016/j.tetlet.2009.06.070
Return to citation in text: [1] -
Tsukamoto, H.; Ueno, T.; Kondo, Y. Org. Lett. 2007, 9, 3033–3036. doi:10.1021/ol071107v
Return to citation in text: [1] -
Ivchenko, P. V.; Nifant’ev, I. E.; Luzikov, Yu. N.; Mkoyan, S. G. Synthesis 2007, 1038–1046. doi:10.1055/s-2007-965957
Return to citation in text: [1] -
Nishikata, T.; Kobayashi, Y.; Kobayshi, K.; Yamamoto, Y.; Miyaura, N. Synlett 2007, 3055–3057. doi:10.1055/s-2007-990964
Return to citation in text: [1] -
Bordwell, F. G.; Drucker, G. E.; Fried, H. E. J. Org. Chem. 1981, 46, 632–635. doi:10.1021/jo00316a032
Return to citation in text: [1] -
Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1991, 56, 4218–4223. doi:10.1021/jo00013a027
Return to citation in text: [1] -
Rathman, T. L.; Bailey, W. F. Org. Process Res. Dev. 2009, 13, 144–151. doi:10.1021/op800246z
Return to citation in text: [1] -
Kelly, P. A.; Berger, G. O.; Wyatt, J. K.; Nantz, M. H. J. Org. Chem. 2003, 68, 8447–8452. doi:10.1021/jo034975o
And references therein.
Return to citation in text: [1] -
Resconi, L.; Piemontesi, F.; Camurati, I.; Balboni, D.; Sironi, A.; Moret, M.; Rychlicki, H.; Zeigler, R. Organometallics 1996, 15, 5046–5059. doi:10.1021/om9604233
Return to citation in text: [1] -
Koelsch, C. F.; LeClaire, C. D. J. Org. Chem. 1941, 6, 516–533. doi:10.1021/jo01204a005
Return to citation in text: [1]
| 17. |
Alcalde, E.; Mesquida, N.; Frigola, J.; López-Pérez, S.; Mercè, R. Org. Biomol. Chem. 2008, 6, 3795–3810. doi:10.1039/b808641a
And references cited therein. |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 7. | Leino, R.; Lehmus, P.; Lehtonen, A. Eur. J. Inorg. Chem. 2004, 3201–3222. doi:10.1002/ejic.200400214 |
| 1. | Chirik, P. J. Organometallics 2010, 29, 1500–1517. doi:10.1021/om100016p |
| 2. | Zhang, C.; Luo, F.; Cheng, B.; Li, B.; Song, H.; Xu, S.; Wang, B. Dalton Trans. 2009, 7230–7235. doi:10.1039/b906933j |
| 3. |
Guo, S.; Liu, Y. Org. Biomol. Chem. 2008, 6, 2064–2070. doi:10.1039/b804888f
And references therein. |
| 4. | Guan, Z.-H.; Ren, Z.-H.; Zhao, L.-B.; Liang, Y.-M. Org. Biomol. Chem. 2008, 6, 1040–1045. doi:10.1039/b719957k |
| 5. | Enders, M.; Baker, R. W. Curr. Org. Chem. 2006, 10, 937–953. doi:10.2174/138527206777435508 |
| 6. | Silver, S.; Leppänen, A.-S.; Sjöholm, R.; Penninkangas, A.; Leino, R. Eur. J. Org. Chem. 2005, 1058–1081. doi:10.1002/ejoc.200400789 |
| 7. | Leino, R.; Lehmus, P.; Lehtonen, A. Eur. J. Inorg. Chem. 2004, 3201–3222. doi:10.1002/ejic.200400214 |
| 8. | Zargarian, D. Coord. Chem. Rev. 2002, 233–234, 157–176. doi:10.1016/S0010-8545(02)00201-1 |
| 9. | Hummel, S.; Kirsch, S. F. Beilstein J. Org. Chem. 2011, 7, 847–859. doi:10.3762/bjoc.7.97 |
| 17. |
Alcalde, E.; Mesquida, N.; Frigola, J.; López-Pérez, S.; Mercè, R. Org. Biomol. Chem. 2008, 6, 3795–3810. doi:10.1039/b808641a
And references cited therein. |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 23. | Bordwell, F. G.; Drucker, G. E.; Fried, H. E. J. Org. Chem. 1981, 46, 632–635. doi:10.1021/jo00316a032 |
| 24. | Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1991, 56, 4218–4223. doi:10.1021/jo00013a027 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 3. |
Guo, S.; Liu, Y. Org. Biomol. Chem. 2008, 6, 2064–2070. doi:10.1039/b804888f
And references therein. |
| 17. |
Alcalde, E.; Mesquida, N.; Frigola, J.; López-Pérez, S.; Mercè, R. Org. Biomol. Chem. 2008, 6, 3795–3810. doi:10.1039/b808641a
And references cited therein. |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 19. | Wang, J.; Zhang, L.; Jing, Y.; Huang, W.; Zhou, X. Tetrahedron Lett. 2009, 50, 4978–4982. doi:10.1016/j.tetlet.2009.06.070 |
| 20. | Tsukamoto, H.; Ueno, T.; Kondo, Y. Org. Lett. 2007, 9, 3033–3036. doi:10.1021/ol071107v |
| 21. | Ivchenko, P. V.; Nifant’ev, I. E.; Luzikov, Yu. N.; Mkoyan, S. G. Synthesis 2007, 1038–1046. doi:10.1055/s-2007-965957 |
| 22. | Nishikata, T.; Kobayashi, Y.; Kobayshi, K.; Yamamoto, Y.; Miyaura, N. Synlett 2007, 3055–3057. doi:10.1055/s-2007-990964 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 10. | Waldmann, H.; Karaguni, I.-M.; Carpintero, M.; Gourzoulidou, E.; Herrmann, C.; Brockmann, C.; Oschkinat, H.; Müller, O. Angew. Chem., Int. Ed. 2004, 43, 454–458. doi:10.1002/anie.200353089 |
| 11. | Karaguni, I.-M.; Glüsenkamp, K.-H.; Langerak, A.; Geisen, C.; Ullrich, V.; Winde, G.; Möröy, T.; Müller, O. Bioorg. Med. Chem. Lett. 2002, 12, 709–713. doi:10.1016/S0960-894X(01)00839-3 |
| 12. | Li, B.-F.; Moree, W. J.; Yu, J.; Coon, T.; Zamani-Kord, S.; Malany, S.; Jalali, K.; Wen, J.; Wang, H.; Yang, C.; Hoare, S. R. J.; Petroski, R. E.; Madan, A.; Crowe, P. D.; Beaton, G. Bioorg. Med. Chem. Lett. 2010, 20, 2629–2633. doi:10.1016/j.bmcl.2010.02.055 |
| 13. | Moree, W. J.; Li, B.-F.; Jovic, F.; Coon, T.; Yu, J.; Gross, R. S.; Tucci, F.; Marinkovic, D.; Zamani-Kord, S.; Malany, S.; Bradbury, M. J.; Hernandez, L. M.; O’Brien, Z.; Wen, J.; Wang, H.; Hoare, S. R. J.; Petroski, R. E.; Sacaan, A.; Madan, A.; Crowe, P. D.; Beaton, G. J. Med. Chem. 2009, 52, 5307–5310. doi:10.1021/jm900933k |
| 14. | Hodgson, D. M.; Winning, L. H. Beilstein J. Org. Chem. 2008, 4, No. 38. doi:10.3762/bjoc.4.38 |
| 15. | Huffman, J. W.; Padgett, L. W. Curr. Med. Chem. 2005, 12, 1395–1411. doi:10.2174/0929867054020864 |
| 16. | Böhme, T. M.; Keim, C.; Kreutzmann, K.; Linder, M.; Dingermann, T.; Dannhardt, G.; Mutschler, E.; Lambrecht, G. J. Med. Chem. 2003, 46, 856–867. doi:10.1021/jm020895l |
| 17. |
Alcalde, E.; Mesquida, N.; Frigola, J.; López-Pérez, S.; Mercè, R. Org. Biomol. Chem. 2008, 6, 3795–3810. doi:10.1039/b808641a
And references cited therein. |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 6. | Silver, S.; Leppänen, A.-S.; Sjöholm, R.; Penninkangas, A.; Leino, R. Eur. J. Org. Chem. 2005, 1058–1081. doi:10.1002/ejoc.200400789 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 25. | Rathman, T. L.; Bailey, W. F. Org. Process Res. Dev. 2009, 13, 144–151. doi:10.1021/op800246z |
| 28. | Koelsch, C. F.; LeClaire, C. D. J. Org. Chem. 1941, 6, 516–533. doi:10.1021/jo01204a005 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
| 26. |
Kelly, P. A.; Berger, G. O.; Wyatt, J. K.; Nantz, M. H. J. Org. Chem. 2003, 68, 8447–8452. doi:10.1021/jo034975o
And references therein. |
| 27. | Resconi, L.; Piemontesi, F.; Camurati, I.; Balboni, D.; Sironi, A.; Moret, M.; Rychlicki, H.; Zeigler, R. Organometallics 1996, 15, 5046–5059. doi:10.1021/om9604233 |
| 18. | Alcalde, E.; Mesquida, N.; López-Pérez, S.; Frigola, J.; Mercè, R. J. Med. Chem. 2009, 52, 675–687. doi:10.1021/jm8009469 |
© 2011 Mesquida et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)