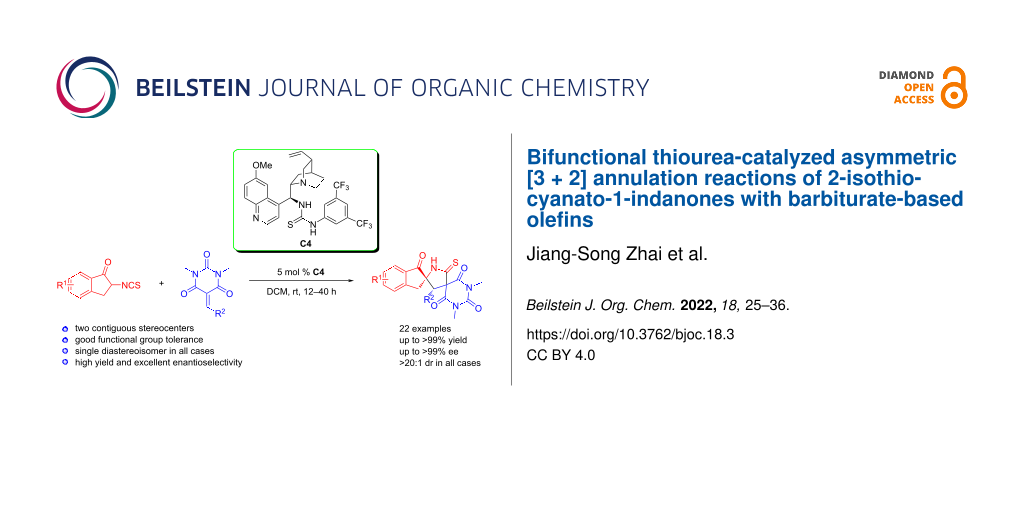
Abstract
Bifunctional thiourea-catalyzed asymmetric [3 + 2] annulation reactions of 2-isothiocyanato-1-indanones with barbiturate-based olefins have been developed to afford chiral dispiro[indene-pyrrolidine-pyrimidine]s. Through this strategy, the target products could be obtained in good to excellent yields with excellent stereoselectivities. In addition, the synthetic utility was verified through a gram-scale synthesis, one-pot three-component reactions and further transformation experiments of the products.

Graphical Abstract
Introduction
Indane scaffolds exist in various biologically active natural products and pharmaceutical compounds with antipsychotic and antifungal activities, such as SB 209670, indatraline, tefludazine, mutisianthol, rasagiline, and ramelteon (Figure 1) [1-5]. Therefore, this structural motif has attracted great attention of researchers in the field of synthetic organic chemistry and pharmaceutical chemistry all over the world. In the previous few decades, a large number of strategies emerged to construct heterocyclic compounds with this skeleton or similar ones [6-10], aiming to explore biological activity and medicinal value conveniently and comprehensively. However, as we know, the construction of these compounds is mostly carried out through transition-metal-catalyzed cyclization reactions [11-14], whereas strategies using bifunctional chiral thiourea catalysts are rarely reported. In 2018, Du's group reported a novel cascade reagent with the indane framework, namely, 2-isothiocyanato-1-indanone (Scheme 1a) [15], but research on its participation in the construction of chiral compounds has been relatively low [16,17].
![[1860-5397-18-3-1]](/bjoc/content/figures/1860-5397-18-3-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of natural products and drugs possessing the indane scaffold.
Figure 1: Selected examples of natural products and drugs possessing the indane scaffold.
![[1860-5397-18-3-i1]](/bjoc/content/inline/1860-5397-18-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Known strategies and conceptual advance of this contribution.
Scheme 1: Known strategies and conceptual advance of this contribution.
On the other hand, as a kind of vital spiroheterocyclic derivatives, spirobarbiturates show a wide range of significant pharmacological and physiological activities in the medical and biological fields (Figure 2) [18-21]. For instance, compound A displays anticonvulsant activity and compound C can be used as an antifungal agent [22,23]. This impels the quest to develop a series of synthons or new methodologies to construct the spirobarbiturates with diverse structures. In recent years, good progress has been achieved in the construction of racemates of spirobarbiturates and the enantioselective synthesis [24-29], but only limited progress has been made in the construction of bispirobarbiturates [30,31]. In 2019, for example, An and co-workers reported an asymmetric Michael/Mannich [3 + 2] cycloaddition reaction between N-(2,2,2-trifluoroethyl)isatin ketimines and barbiturate-based olefins (Scheme 1a) [32]. Based on the current knowledge, the construction of dispirobarbiturates containing the indane skeleton has not been reported yet.
![[1860-5397-18-3-2]](/bjoc/content/figures/1860-5397-18-3-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected examples of bioactive spirobarbiturates.
Figure 2: Selected examples of bioactive spirobarbiturates.
In light of the prominent bioactivities and the pharmacological activity of the above two framework compounds, the combination of these two species may be potential drug candidates. Therefore, it is of great significance to develop a new strategy to construct a series of spirobarbiturates derived from indanone. Combining current researches of these two compounds, we report the first organocatalytic asymmetric Michael addition/cyclization reaction between barbituric acid-derived olefins and indanones (Scheme 1b). Under the action of the bifunctional thiourea catalyst, a series of target products in excellent yields with excellent stereoselectivities can be obtained under mild conditions in this reaction. Notably, this protocol provides direct access to indanone-derived spirobarbiturates, which are difficult to access with other methods.
Results and Discussion
To verify the feasibility of the reaction, the domino Michael addition/cyclization reaction of 2-isothiocyanato-1-indanone (1a) and barbiturate-based olefin 2a was used as a model reaction, which was carried out in dichloromethane (DCM) with 5 mol % quinine-derived squaramide C1 at room temperature. The results are summarized in Table 1. We were pleased to find that the domino Michael addition/cyclization reaction could complete in the presence of 5 mol % C1 at room temperature in 12 h providing the desired product 3aa in 55% yield with excellent stereoselectivity (>20:1 dr, 97% ee) (Table 1, entry 1). Due to the excellent stereoselectivity of the target product 3aa, the reaction conditions were further optimized to increase its yield.
Table 1: Optimization of the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-3-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Catalyst | Yieldb (%) | drc | eed (%) |
| 1 | DCM | C1 | 55 | >20:1 | 97 |
| 2 | DCM | C2 | 57 | >20:1 | 89 |
| 3 | DCM | C3 | 61 | >20:1 | 97 |
| 4 | DCM | C4 | 81 | >20:1 | 97 |
| 5 | DCM | C5 | 79 | >20:1 | 96 |
| 6 | DCM | C6 | 65 | >20:1 | 94 |
| 7 | DCM | C7 | 84 | >20:1 | 94 |
| 8 | DCM | C8 | 82 | >20:1 | 96 |
| 9 | CHCl3 | C4 | 83 | >20:1 | 94 |
| 10 | PhMe | C4 | 80 | >20:1 | 95 |
| 11 | THF | C4 | 81 | >20:1 | 86 |
| 12 | MeCN | C4 | 79 | >20:1 | 94 |
| 13 | DCE | C4 | 72 | >20:1 | 96 |
| 14 | dioxane | C4 | 81 | >20:1 | 96 |
| 15 | EtOAc | C4 | 74 | >20:1 | 93 |
| 16e | DCM | C4 | 76 | >20:1 | 94 |
aUnless otherwise specified, the reactions were carried out with 1a (0.12 mmol), 2a (0.10 mmol) and catalyst (5 mol %) in solvent (1.0 mL) at room temperature for 12 h. bIsolated yield after column chromatography purification. cDetermined by 1H NMR analysis. dEnantiomeric excess (ee) was determined by HPLC analysis. e2.5 mol % catalyst was used and reaction time was 18 h.
Subsequently, a number of organocatalysts (Figure 3) were evaluated for this domino process (Table 1, entries 2–8). From the experimental results, it was found that the yield of product did not increase significantly with the cinchona alkaloid-derived squaramide catalysts (Table 1, entries 2 and 3). Consequently, we decided to explore the effects of different types of catalysts on the reaction. Through experiments, it can be found that thiourea catalysts (C4–C7) can catalyze the reaction to obtain higher yields while keeping the stereoselectivity basically unchanged. Then we chose the C4 catalyst with the best reaction effect as the optimal catalyst to explore the influence of other reaction conditions such as solvent type and catalyst loading on the reaction (Table 1, entries 9–16). The experimental results show that the solvent has a non-negligible effect on the reaction and dichloromethane (DCM) has the best reaction effect in the annulation system (Table 1, entries 9–15). Hereafter, we tried to reduce the catalyst loading to further improve the reaction yield and enantioselectivity, but it did not meet our expectations (Table 1, entry 16). Taking into account the ease of operation of the experiment and for economic reasons, we did not explore the effect of increasing the catalyst loading and changing the reaction temperature on the reaction. Based on the above evaluation, we finally selected 2-isothiocyanato-1-indanones 1 and barbiturate-based olefins 2 with a molar ratio of 1.2:1 to react for 12 h at room temperature in DCM using 5 mol % of catalyst C4 as the optimum reaction conditions.
![[1860-5397-18-3-3]](/bjoc/content/figures/1860-5397-18-3-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
With the optimum reaction conditions established, we then commenced to probe the substrate scope and limitations of this reaction. As summarized in Scheme 2, a variety of 2-isothiocyanato-1-indanones 1 were firstly tested under the optimized conditions. When a methyl substituent is located at the 6-position of the indanone, the reaction yield of the product 3ba was higher than that of the model reaction, but the enantioselectivity was partially reduced. When the 5-position of the indanone was substituted by either F or a MeO group, the yield remained nearly unchanged, however, the enantioselectivity was slightly reduced. On the other hand, when the 5-position of the indanone was substituted by Br, the 6-position was substituted by a MeO group, and the 5 and 6-positions are simultaneously substituted by a MeO group, the yields and stereoselectivities of the reactions significantly dropped. This indicates that the position of the substituent has a great influence on the reaction. Gratifyingly, the diastereoselectivities of the reactions were maintained.
![[1860-5397-18-3-i2]](/bjoc/content/inline/1860-5397-18-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of 2-isothiocyanato-1-indanones. The reactions were carried out with 1 (0.12 mmol), 2a (0.10 mmol), and catalyst (5 mol %) in solvent (1.0 mL) at room temperature for 12 h. The yields refer to the isolated products after column chromatography. The diastereoisomeric ratios (dr values) were determined by 1H NMR spectroscopy and the enantiomeric excess (ee) values were determined by HPLC analysis.
Scheme 2: Substrate scope of 2-isothiocyanato-1-indanones. The reactions were carried out with 1 (0.12 mmol), ...
To further explore the generality of this reaction, structurally diverse barbiturate-based olefins 2 were examined under the standard conditions by reacting with 1a. As shown in Scheme 3, in addition to substrates 3ae and 3al, it appeared that the reaction could well tolerated the presence of electron-donating and electron-withdrawing groups on the benzene ring of substrates 2, and afforded most of the products 3 in excellent chemical yields (90 to >99%) and stereoselectivities (>20:1 dr, 84–>99% ee). Possibly due to the influence of steric hindrance, reactions involving substrate 2 with ortho-substitution on the benzene ring has lower yields and worse enantioselectivities than those with meta-substitution and para-substitution. Meanwhile, the enantioselectivities of the products 3am and 3ao were partially decreased when the R1 group was substituted by naphthyl and thienyl, respectively. It was a good result that R1 was substituted by furyl. Unfortunately, when the R1 was a pyridyl group, the product was obtained in trace amounts. This may be partly related to the poor solubility of this substrate.
![[1860-5397-18-3-i3]](/bjoc/content/inline/1860-5397-18-3-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of barbiturate-based olefins. The reactions were carried out with 1a (0.12 mmol), 2 (0.10 mmol) and catalyst C4 (5 mol %) in solvent (1.0 mL) at room temperature for 12–40 h. The yields refer to isolated products after column chromatography. The diastereoisomeric ratios (dr values) were determined by 1H NMR spectroscopy and the enantiomeric excess (ee) values were determined by HPLC analysis.
Scheme 3: Substrate scope of barbiturate-based olefins. The reactions were carried out with 1a (0.12 mmol), 2...
The absolute configuration of the chiral product 3ae was unambiguously identified on the basis of single-crystal X-ray diffraction analysis as (2S,3'S) (Figure 4) [33]. The configurations of the other products were assigned by analogy to 3ae.
![[1860-5397-18-3-4]](/bjoc/content/figures/1860-5397-18-3-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of 3ae (displacement ellipsoids are drawn at the 50% probability level).
Figure 4: X-ray crystal structure of 3ae (displacement ellipsoids are drawn at the 50% probability level).
In order to further prove the application value of this asymmetric domino Michael addition/cyclization reaction, a gram-scale experiment was performed under the optimized conditions. As exemplified in Scheme 4, the desired dispiro[indene-pyrrolidine-pyrimidine] 3ah could be obtained in 94% yield with excellent stereoselectivity (>20:1 dr, >99% ee), which indicated this strategy shows promising prospects for mass production.
![[1860-5397-18-3-i4]](/bjoc/content/inline/1860-5397-18-3-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Moreover, two different transformations of the product 3ah are shown to validate synthetic utility of the reaction. As demonstrated in Scheme 5, the dispiro[indene-pyrrolidine-pyrimidine] 3ah could be easily oxidized to compound 4 with m-chloroperbenzoic acid under mild conditions, and compound 4 can basically maintain the original excellent stereoselectivity (Scheme 5a). Meanwhile, we are pleased that methylation of 3ah took place easily to afford product 5 in 95% chemical yield with 99% ee and >20:1 dr under the basic reaction conditions (Scheme 5b).
![[1860-5397-18-3-i5]](/bjoc/content/inline/1860-5397-18-3-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
A one-pot reaction of three available starting materials was tested using CH2Cl2 as the solvent. The one-pot reaction of 1,3-dimethylbarbituric acid (6), benzaldehyde (7), and 2-isothiocyanato-1-indanone (1a) proceeded smoothly to provide the desired product 3aa in 80% yield with 95% ee and >20:1 dr (Scheme 6a). In addition, the one-pot reaction of 1,3-dimethylbarbituric acid (6), m-bromobenzaldehyde (8), and 2-isothiocyanato-1-indanone (1a) was also investigated, and the reaction yield (80%) was lower than before, but the stereoselectivity (>20:1 dr, >99% ee) could still be maintained (Scheme 6b). This one-pot three-component reaction would be more convenient for potential industrial applications.
![[1860-5397-18-3-i6]](/bjoc/content/inline/1860-5397-18-3-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot three-component reaction.
Scheme 6: One-pot three-component reaction.
Finally, in order to understand the enantioselective formation process of product 3, we proposed the possible mechanisms for the [3 + 2] cyclization reaction of 1a and 2a based on the previously published work. As illustrated in Scheme 7, in this cycle, it is reasonable that the catalyst C4 activates barbiturate-based olefins 2a through the action of hydrogen bonds, and then the 2-isothiocyanato-1-indanone 1a tautomerizes to form the corresponding enol under the action of catalyst C4. Simultaneously, deprotonated 1a attacks the double bond of 2a from the Si face via intermediate A, resulting in a Michael addition reaction. Then the electron-deficient isothiocyanate moiety is attacked by newly generated α-carbon center from barbiturate-based olefins 2a to form intermediate B. Finally, the catalyst C4 is removed in intermediate C and the product 3aa is obtained.
![[1860-5397-18-3-i7]](/bjoc/content/inline/1860-5397-18-3-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have successfully developed an exceptionally efficient strategy for the enantioselective construction of indanone-derived spirobarbiturates through a simple organocatalytic domino Michael/cyclization reaction. This annulation reaction can be easily performed under air atmosphere and mild conditions with 5 mol % catalyst loading. By using bifunctional thiourea catalyst, a series of structurally diverse indanone-derived spirobarbiturates could be obtained in high yields and excellent diastereo- and enantioselectivities (up to >99% yield, >20:1 dr and >99% ee). In addition, a gram-scale synthesis, one-pot three-component reactions and further transformation experiments of the products were also demonstrated with excellent stereoselectivities. We believe that the availability of these compounds will provide promising candidates for chemical biology and drug discovery.
Experimental
General information
Commercially available compounds were used without further purification. Solvents were dried according to standard procedures. Column chromatography was performed with silica gel (200–300 mesh). Melting points were determined with an XT-4 melting-point apparatus and are uncorrected. 1H NMR spectra were measured with a Bruker Ascend 400 MHz spectrometer, chemical shifts are reported in δ (ppm) units relative to tetramethylsilane (TMS) as an internal standard. 13C NMR spectra were measured at 100 MHz with a 400 MHz spectrometer or at 176 MHz with a 700 MHz spectrometer, chemical shifts are reported in δ (ppm) units relative to tetramethylsilane and referenced to solvent peak (CDCl3, δ = 77.00 ppm; DMSO-d6, δ = 39.43 ppm). High-resolution mass spectra were measured with an Agilent 6520 Accurate-Mass Q-TOF MS system equipped with an electrospray ionization (ESI) source. Optical rotations were measured with a Krüss P8000 polarimeter at the indicated concentration with the units of g/100 mL. Enantiomeric excesses were determined by chiral HPLC analysis using an Agilent 1200 LC instrument with a Daicel Chiralpak IB, IC, or ADH column.
The following compounds were prepared following procedures reported in the literature: 1a‒g [15], 2a‒o [34], and chiral organocatalysts [35-38].
1. Procedure for the synthesis of racemates of 3
To a dried small bottle were added 1 (0.06 mmol), 2 (0.05 mmol), Et3N (1.0 mg, 0.01 mmol, 0.2 equiv), and DCM (1.0 mL). The mixture was stirred at room temperature for 12 h, then the reaction mixture was concentrated and directly purified by silica gel column chromatography to afford the racemates of 3.
2. Procedure for the synthesis of chiral compounds 3
To a dried small bottle were added 1 (0.12 mmol), 2 (0.10 mmol), chiral organocatalyst C4 (2.7 mg, 0.005 mmol, 5 mol %), and DCM (1.0 mL). The mixture was stirred at room temperature for 12‒40 h, then the reaction mixture was concentrated and directly purified by silica gel column chromatography to afford the desired products 3.
3. Gram-scale synthesis of 3ah
2-Isothiocyanato-2,3-dihydro-1H-inden-1-one (1a, 1.022 g, 5.4 mmol), 1,3-dimethyl-5-(3-methylbenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (2h, 1.162 g, 4.5 mmol), and catalyst C4 (122.4 mg, 5 mol %) were dissolved in dry DCM (45 mL) at room temperature. After stirring at room temperature for 39 h, the reaction mixture was concentrated, and directly purified by silica gel column chromatography (dichloromethane/ethyl acetate/petroleum ether 1:1:5) to afford the desired product 3ah as white solid (1.976 g, 94% yield) with >20:1 dr and >99% ee.
4. Synthetic procedure for compound 4
The synthesis of compound 4 was similar to the reported method in the literature [39]. In a 5 mL small bottle, compound 3ah (44.8 mg, 0.10 mmol, 1.0 equiv) was dissolved in CH2Cl2 (2 mL) and the bottle was placed in an ice-water bath. Then m-CPBA (≈85%, 60.9 mg, 0.30 mmol, 3.0 equiv) was added to the reaction mixture at 0 °C. After completion of the addition, the reaction mixture was slowly warmed to room temperature and allowed to stir overnight. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate 2:1) to give pure compound 4 as a white solid (35.1 mg, 81% yield).
5. Synthetic procedure for compound 5
The synthesis of compound 5 was similar to the reported method in the literature [39]. To an oven dried 5 mL small bottle were added compound 3ah (44.8 mg, 0.10 mmol, 1.0 equiv), dry K2CO3 (21.0 mg, 0.23 mmol, 1.50 equiv), and THF (2 mL). The solution was cooled to 0 °C and iodomethane (12.5 μL, 0.20 mmol, 2.0 equiv) was added dropwise to the reaction mixture. After completion of the addition, the reaction mixture was gradually warmed to room temperature and allowed to stir overnight. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 4:1) to give pure compound 5 as white solid (44.0 mg, 95% yield).
6. One-pot three-component reaction for the synthesis of 3aa
1,3-Dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (15.6 mg, 0.10 mmol) and benzaldehyde (10.6 mg, 0.10 mmol) were dissolved in anhydrous CH2Cl2 (1.0 mL) and stirred at room temperature for 10 h. Then, catalyst C4 (2.7 mg, 5 mol %) and compound 1a (22.7 mg, 0.12 mmol) were added. After stirring at room temperature for another 12 h, the reaction mixture was concentrated and directly purified by silica gel column chromatography (dichloromethane/ethyl acetate/petroleum ether 1:1:5) to afford the desired product 3aa as white solid (35.5 mg, 80% yield) with >20:1 dr and 95% ee.
7. One-pot three-component reaction for the synthesis of 3ag
1,3-Dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (15.6 mg, 0.10 mmol) and m-bromobenzaldehyde (18.5 mg, 0.10 mmol) were dissolved in anhydrous CH2Cl2 (1.0 mL) and stirred at room temperature for 10 h. Then, catalyst C4 (2.7 mg, 5 mol %) and compound 1a (22.7 mg, 0.12 mmol) were added. After stirring at room temperature for another 12 h, the reaction mixture was concentrated and directly purified by silica gel column chromatography (dichloromethane/ethyl acetate/petroleum ether 1:1:5) to afford the desired product 3ag as a white solid (41.0 mg, 80% yield) with >20:1 dr and >99% ee.
References
-
Svendsen, O.; Arnt, J.; Boeck, V.; Bøgesø, K. P.; Christensen, A. V.; Hyttel, J.; Larsen, J.-J. Drug Dev. Res. 1986, 7, 35–47. doi:10.1002/ddr.430070104
Return to citation in text: [1] -
Bianco, G. G.; Ferraz, H. M. C.; Costa, A. M.; Costa-Lotufo, L. V.; Pessoa, C.; de Moraes, M. O.; Schrems, M. G.; Pfaltz, A.; Silva, L. F., Jr. J. Org. Chem. 2009, 74, 2561–2566. doi:10.1021/jo9000405
Return to citation in text: [1] -
Miyauchi, Y.; Jesmin, S.; Sakai, S.; Kamiyama, J.; Shimojo, N.; Rahman, A.; Islam, M.; Zaedi, S.; Maeda, S.; Maruyama, H.; Mizutani, T.; Homma, S.; Aonuma, K.; Miyauchi, T. Life Sci. 2014, 111, 6–11. doi:10.1016/j.lfs.2014.06.008
Return to citation in text: [1] -
Borie, C.; Ackermann, L.; Nechab, M. Chem. Soc. Rev. 2016, 45, 1368–1386. doi:10.1039/c5cs00622h
Return to citation in text: [1] -
Decker, A. M.; Blough, B. E. J. Pharmacol. Toxicol. Methods 2018, 92, 52–56. doi:10.1016/j.vascn.2018.03.003
Return to citation in text: [1] -
Mizuta, S.; Tsuzuki, T.; Fujimoto, T.; Yamamoto, I. Org. Lett. 2005, 7, 3633–3635. doi:10.1021/ol051129m
Return to citation in text: [1] -
Gabriele, B.; Mancuso, R.; Veltri, L. Chem. – Eur. J. 2016, 22, 5056–5094. doi:10.1002/chem.201503933
Return to citation in text: [1] -
Feng, Z.; Yuan, Z.; Zhao, X.; Huang, Y.; Yao, H. Org. Chem. Front. 2019, 6, 3535–3539. doi:10.1039/c9qo00977a
Return to citation in text: [1] -
Prasher, P.; Sharma, M. ChemistrySelect 2021, 6, 2658–2677. doi:10.1002/slct.202100177
Return to citation in text: [1] -
Laina‐Martín, V.; Fernández‐Salas, J. A.; Alemán, J. Chem. – Eur. J. 2021, 27, 12509–12520. doi:10.1002/chem.202101696
Return to citation in text: [1] -
Hu, J.; Hirao, H.; Li, Y.; Zhou, J. S. Angew. Chem., Int. Ed. 2013, 52, 8676–8680. doi:10.1002/anie.201303753
Return to citation in text: [1] -
Yue, G.; Lei, K.; Hirao, H.; Zhou, J. S. Angew. Chem., Int. Ed. 2015, 54, 6531–6535. doi:10.1002/anie.201501712
Return to citation in text: [1] -
Matsuda, T.; Watanuki, S. Org. Biomol. Chem. 2015, 13, 702–705. doi:10.1039/c4ob02210f
Return to citation in text: [1] -
Léonard, N. G.; Palmer, W. N.; Friedfeld, M. R.; Bezdek, M. J.; Chirik, P. J. ACS Catal. 2019, 9, 9034–9044. doi:10.1021/acscatal.9b03444
Return to citation in text: [1] -
Zhao, B.-L.; Du, D.-M. Org. Lett. 2018, 20, 3797–3800. doi:10.1021/acs.orglett.8b01389
Return to citation in text: [1] [2] -
Hou, X.-Q.; Lin, Y.; Du, D.-M. Org. Chem. Front. 2021, 8, 4183–4187. doi:10.1039/d1qo00626f
Return to citation in text: [1] -
Hou, X.-Q.; Wen, J.-B.; Yan, L.; Du, D.-M. Org. Biomol. Chem. 2021, 19, 7181–7185. doi:10.1039/d1ob01223a
Return to citation in text: [1] -
Kim, S.-H.; Pudzianowski, A. T.; Leavitt, K. J.; Barbosa, J.; McDonnell, P. A.; Metzler, W. J.; Rankin, B. M.; Liu, R.; Vaccaro, W.; Pitts, W. Bioorg. Med. Chem. Lett. 2005, 15, 1101–1106. doi:10.1016/j.bmcl.2004.12.016
Return to citation in text: [1] -
Duan, J. J.-W.; Chen, L.; Lu, Z.; Jiang, B.; Asakawa, N.; Sheppeck, J. E., II; Liu, R.-Q.; Covington, M. B.; Pitts, W.; Kim, S.-H.; Decicco, C. P. Bioorg. Med. Chem. Lett. 2007, 17, 266–271. doi:10.1016/j.bmcl.2006.09.048
Return to citation in text: [1] -
Bhaskarachar, R. K.; Revanasiddappa, V. G.; Hegde, S.; Balakrishna, J. P.; Reddy, S. Y. Med. Chem. Res. 2015, 24, 3516–3528. doi:10.1007/s00044-015-1408-7
Return to citation in text: [1] -
Xu, H.; Huang, R.-L.; Shu, Z.; Hong, R.; Zhang, Z. Org. Biomol. Chem. 2021, 19, 4978–4985. doi:10.1039/d1ob00508a
Return to citation in text: [1] -
Galati, E. M.; Monforte, M. T.; Miceli, N.; Raneri, E. Farmaco 2001, 56, 459–461. doi:10.1016/s0014-827x(01)01062-x
Return to citation in text: [1] -
Ingle, V. N.; Gaidhane, P. K.; Dutta, S. S.; Naha, P. P.; Sengupta, M. S. J. Carbohydr. Chem. 2006, 25, 661–671. doi:10.1080/07328300601039328
Return to citation in text: [1] -
Renard, A.; Lhomme, J.; Kotera, M. J. Org. Chem. 2002, 67, 1302–1307. doi:10.1021/jo016194y
Return to citation in text: [1] -
Lomlim, L.; Einsiedel, J.; Heinemann, F. W.; Meyer, K.; Gmeiner, P. J. Org. Chem. 2008, 73, 3608–3611. doi:10.1021/jo702573z
Return to citation in text: [1] -
Ramachary, D. B.; Reddy, Y. V.; Banerjee, A.; Banerjee, S. Org. Biomol. Chem. 2011, 9, 7282–7286. doi:10.1039/c1ob06133j
Return to citation in text: [1] -
Hosseini, Y.; Rastgar, S.; Heren, Z.; Büyükgüngörc, O.; Pesyan, N. N. J. Chin. Chem. Soc. 2011, 58, 309–318. doi:10.1002/jccs.201190031
Return to citation in text: [1] -
Huang, Z.; Zhao, Q.; Chen, G.; Wang, H.; Lin, W.; Xu, L.; Liu, H.; Wang, J.; Shi, D.; Wang, Y. Molecules 2012, 17, 12704–12717. doi:10.3390/molecules171112704
Return to citation in text: [1] -
Liu, H.; Liu, Y.; Yuan, C.; Wang, G.-P.; Zhu, S.-F.; Wu, Y.; Wang, B.; Sun, Z.; Xiao, Y.; Zhou, Q.-L.; Guo, H. Org. Lett. 2016, 18, 1302–1305. doi:10.1021/acs.orglett.6b00239
Return to citation in text: [1] -
Zhao, H.-W.; Tian, T.; Li, B.; Yang, Z.; Pang, H.-L.; Meng, W.; Song, X.-Q.; Chen, X.-Q. J. Org. Chem. 2015, 80, 10380–10385. doi:10.1021/acs.joc.5b01810
Return to citation in text: [1] -
Zhao, H.-W.; Tian, T.; Pang, H.-L.; Li, B.; Chen, X.-Q.; Yang, Z.; Meng, W.; Song, X.-Q.; Zhao, Y.-D.; Liu, Y.-Y. Adv. Synth. Catal. 2016, 358, 2619–2630. doi:10.1002/adsc.201600270
Return to citation in text: [1] -
An, T.-L.; Du, D.-M. ChemistrySelect 2019, 4, 11302–11306. doi:10.1002/slct.201903146
Return to citation in text: [1] -
CCDC 2107568 (for 3ae) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Return to citation in text: [1] -
Liu, Y.; Yang, W.; Wu, Y.; Mao, B.; Gao, X.; Liu, H.; Sun, Z.; Xiao, Y.; Guo, H. Adv. Synth. Catal. 2016, 358, 2867–2872. doi:10.1002/adsc.201600450
Return to citation in text: [1] -
Zhu, Y.; Malerich, J. P.; Rawal, V. H. Angew. Chem., Int. Ed. 2010, 49, 153–156. doi:10.1002/anie.200904779
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Adv. Synth. Catal. 2011, 353, 1241–1246. doi:10.1002/adsc.201000981
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] -
Lin, Y.; Liu, L.; Du, D.-M. Org. Chem. Front. 2017, 4, 1229–1238. doi:10.1039/c6qo00852f
Return to citation in text: [1] [2]
| 1. | Svendsen, O.; Arnt, J.; Boeck, V.; Bøgesø, K. P.; Christensen, A. V.; Hyttel, J.; Larsen, J.-J. Drug Dev. Res. 1986, 7, 35–47. doi:10.1002/ddr.430070104 |
| 2. | Bianco, G. G.; Ferraz, H. M. C.; Costa, A. M.; Costa-Lotufo, L. V.; Pessoa, C.; de Moraes, M. O.; Schrems, M. G.; Pfaltz, A.; Silva, L. F., Jr. J. Org. Chem. 2009, 74, 2561–2566. doi:10.1021/jo9000405 |
| 3. | Miyauchi, Y.; Jesmin, S.; Sakai, S.; Kamiyama, J.; Shimojo, N.; Rahman, A.; Islam, M.; Zaedi, S.; Maeda, S.; Maruyama, H.; Mizutani, T.; Homma, S.; Aonuma, K.; Miyauchi, T. Life Sci. 2014, 111, 6–11. doi:10.1016/j.lfs.2014.06.008 |
| 4. | Borie, C.; Ackermann, L.; Nechab, M. Chem. Soc. Rev. 2016, 45, 1368–1386. doi:10.1039/c5cs00622h |
| 5. | Decker, A. M.; Blough, B. E. J. Pharmacol. Toxicol. Methods 2018, 92, 52–56. doi:10.1016/j.vascn.2018.03.003 |
| 16. | Hou, X.-Q.; Lin, Y.; Du, D.-M. Org. Chem. Front. 2021, 8, 4183–4187. doi:10.1039/d1qo00626f |
| 17. | Hou, X.-Q.; Wen, J.-B.; Yan, L.; Du, D.-M. Org. Biomol. Chem. 2021, 19, 7181–7185. doi:10.1039/d1ob01223a |
| 39. | Lin, Y.; Liu, L.; Du, D.-M. Org. Chem. Front. 2017, 4, 1229–1238. doi:10.1039/c6qo00852f |
| 15. | Zhao, B.-L.; Du, D.-M. Org. Lett. 2018, 20, 3797–3800. doi:10.1021/acs.orglett.8b01389 |
| 39. | Lin, Y.; Liu, L.; Du, D.-M. Org. Chem. Front. 2017, 4, 1229–1238. doi:10.1039/c6qo00852f |
| 11. | Hu, J.; Hirao, H.; Li, Y.; Zhou, J. S. Angew. Chem., Int. Ed. 2013, 52, 8676–8680. doi:10.1002/anie.201303753 |
| 12. | Yue, G.; Lei, K.; Hirao, H.; Zhou, J. S. Angew. Chem., Int. Ed. 2015, 54, 6531–6535. doi:10.1002/anie.201501712 |
| 13. | Matsuda, T.; Watanuki, S. Org. Biomol. Chem. 2015, 13, 702–705. doi:10.1039/c4ob02210f |
| 14. | Léonard, N. G.; Palmer, W. N.; Friedfeld, M. R.; Bezdek, M. J.; Chirik, P. J. ACS Catal. 2019, 9, 9034–9044. doi:10.1021/acscatal.9b03444 |
| 34. | Liu, Y.; Yang, W.; Wu, Y.; Mao, B.; Gao, X.; Liu, H.; Sun, Z.; Xiao, Y.; Guo, H. Adv. Synth. Catal. 2016, 358, 2867–2872. doi:10.1002/adsc.201600450 |
| 6. | Mizuta, S.; Tsuzuki, T.; Fujimoto, T.; Yamamoto, I. Org. Lett. 2005, 7, 3633–3635. doi:10.1021/ol051129m |
| 7. | Gabriele, B.; Mancuso, R.; Veltri, L. Chem. – Eur. J. 2016, 22, 5056–5094. doi:10.1002/chem.201503933 |
| 8. | Feng, Z.; Yuan, Z.; Zhao, X.; Huang, Y.; Yao, H. Org. Chem. Front. 2019, 6, 3535–3539. doi:10.1039/c9qo00977a |
| 9. | Prasher, P.; Sharma, M. ChemistrySelect 2021, 6, 2658–2677. doi:10.1002/slct.202100177 |
| 10. | Laina‐Martín, V.; Fernández‐Salas, J. A.; Alemán, J. Chem. – Eur. J. 2021, 27, 12509–12520. doi:10.1002/chem.202101696 |
| 35. | Zhu, Y.; Malerich, J. P.; Rawal, V. H. Angew. Chem., Int. Ed. 2010, 49, 153–156. doi:10.1002/anie.200904779 |
| 36. | Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g |
| 37. | Yang, W.; Du, D.-M. Adv. Synth. Catal. 2011, 353, 1241–1246. doi:10.1002/adsc.201000981 |
| 38. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 30. | Zhao, H.-W.; Tian, T.; Li, B.; Yang, Z.; Pang, H.-L.; Meng, W.; Song, X.-Q.; Chen, X.-Q. J. Org. Chem. 2015, 80, 10380–10385. doi:10.1021/acs.joc.5b01810 |
| 31. | Zhao, H.-W.; Tian, T.; Pang, H.-L.; Li, B.; Chen, X.-Q.; Yang, Z.; Meng, W.; Song, X.-Q.; Zhao, Y.-D.; Liu, Y.-Y. Adv. Synth. Catal. 2016, 358, 2619–2630. doi:10.1002/adsc.201600270 |
| 33. | CCDC 2107568 (for 3ae) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. |
| 24. | Renard, A.; Lhomme, J.; Kotera, M. J. Org. Chem. 2002, 67, 1302–1307. doi:10.1021/jo016194y |
| 25. | Lomlim, L.; Einsiedel, J.; Heinemann, F. W.; Meyer, K.; Gmeiner, P. J. Org. Chem. 2008, 73, 3608–3611. doi:10.1021/jo702573z |
| 26. | Ramachary, D. B.; Reddy, Y. V.; Banerjee, A.; Banerjee, S. Org. Biomol. Chem. 2011, 9, 7282–7286. doi:10.1039/c1ob06133j |
| 27. | Hosseini, Y.; Rastgar, S.; Heren, Z.; Büyükgüngörc, O.; Pesyan, N. N. J. Chin. Chem. Soc. 2011, 58, 309–318. doi:10.1002/jccs.201190031 |
| 28. | Huang, Z.; Zhao, Q.; Chen, G.; Wang, H.; Lin, W.; Xu, L.; Liu, H.; Wang, J.; Shi, D.; Wang, Y. Molecules 2012, 17, 12704–12717. doi:10.3390/molecules171112704 |
| 29. | Liu, H.; Liu, Y.; Yuan, C.; Wang, G.-P.; Zhu, S.-F.; Wu, Y.; Wang, B.; Sun, Z.; Xiao, Y.; Zhou, Q.-L.; Guo, H. Org. Lett. 2016, 18, 1302–1305. doi:10.1021/acs.orglett.6b00239 |
| 15. | Zhao, B.-L.; Du, D.-M. Org. Lett. 2018, 20, 3797–3800. doi:10.1021/acs.orglett.8b01389 |
| 22. | Galati, E. M.; Monforte, M. T.; Miceli, N.; Raneri, E. Farmaco 2001, 56, 459–461. doi:10.1016/s0014-827x(01)01062-x |
| 23. | Ingle, V. N.; Gaidhane, P. K.; Dutta, S. S.; Naha, P. P.; Sengupta, M. S. J. Carbohydr. Chem. 2006, 25, 661–671. doi:10.1080/07328300601039328 |
| 18. | Kim, S.-H.; Pudzianowski, A. T.; Leavitt, K. J.; Barbosa, J.; McDonnell, P. A.; Metzler, W. J.; Rankin, B. M.; Liu, R.; Vaccaro, W.; Pitts, W. Bioorg. Med. Chem. Lett. 2005, 15, 1101–1106. doi:10.1016/j.bmcl.2004.12.016 |
| 19. | Duan, J. J.-W.; Chen, L.; Lu, Z.; Jiang, B.; Asakawa, N.; Sheppeck, J. E., II; Liu, R.-Q.; Covington, M. B.; Pitts, W.; Kim, S.-H.; Decicco, C. P. Bioorg. Med. Chem. Lett. 2007, 17, 266–271. doi:10.1016/j.bmcl.2006.09.048 |
| 20. | Bhaskarachar, R. K.; Revanasiddappa, V. G.; Hegde, S.; Balakrishna, J. P.; Reddy, S. Y. Med. Chem. Res. 2015, 24, 3516–3528. doi:10.1007/s00044-015-1408-7 |
| 21. | Xu, H.; Huang, R.-L.; Shu, Z.; Hong, R.; Zhang, Z. Org. Biomol. Chem. 2021, 19, 4978–4985. doi:10.1039/d1ob00508a |
| 32. | An, T.-L.; Du, D.-M. ChemistrySelect 2019, 4, 11302–11306. doi:10.1002/slct.201903146 |
© 2022 Zhai and Du; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.