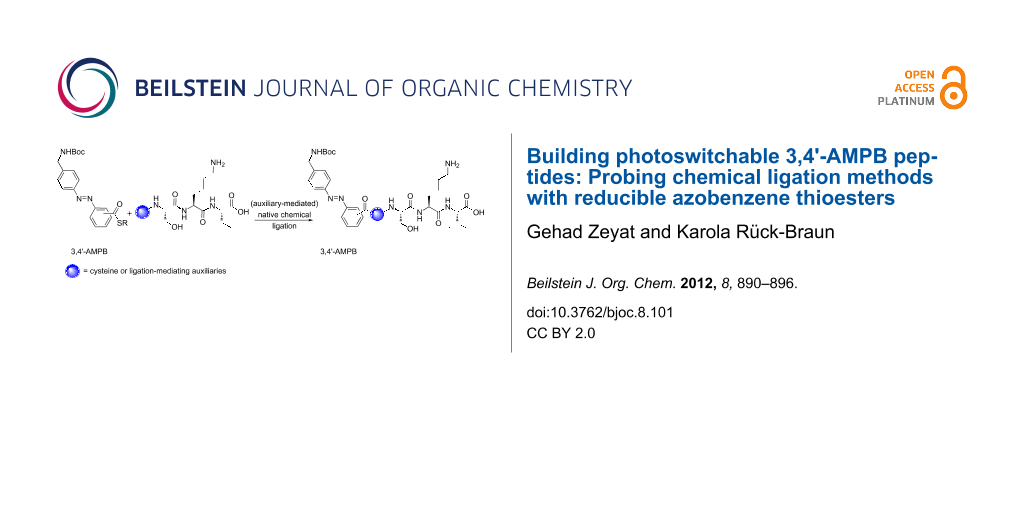
Abstract
Photoswitchable peptides were synthesized by using cysteine- and auxiliary-based native chemical ligation reactions. For this purpose, the two regioisomeric azobenzene building blocks 3,4'-AMPB thioester 1b and 4,4'-AMPB thioester 2b were employed in the ligation reactions. While 4,4'-AMPB requires the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary to minimize reduction of the diazene unit, 3,4'-AMPB can be used in combination with the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary as well as the Nα-2-mercaptoethyl auxiliary. Thus, 3,4'-AMPB derivatives/peptides proved to be significantly less prone to reduction by aliphatic and aromatic thiols than were the 4,4'-AMPB compounds.

Graphical Abstract
Introduction
Optical switches not only offer the advantage to elucidate, but also to control biological processes with high spatial and temporal resolution by using light, either in vitro or in vivo [1]. In this context, azobenzenes remain a privileged class of photoswitches, and typical applications based on light-triggered reversible conformational control of cyclic, helical or beta-hairpin peptides have been intensively reviewed [2,3]. Current research in this field focuses on advanced methods and designs for the synthesis of complex azopeptides and azoproteins for ambitious biophysical studies, and also for intracellular applications. In this respect, an increased and predictable stability of azobenzene building blocks under reducing conditions seems to be a prerequisite in light of synthetic challenges, but also when considering the reducing intracellular environment. We recently reported on the synthesis, properties and applications of a series of novel azobenzene ω-amino acids, with a preference for meta-substitution patterns. Our purpose was amongst other things (i) to increase flexibility, and (ii) to suppress resonance effects in order to enhance the stability of the diazene unit [4,5]. Since methods based on native chemical ligation (NCL), e.g., NCL with Nα-acyl transfer auxiliaries, have gained considerable interest in the past two decades [6], we reasoned that a comparative ligation study with the Boc-protected azobenzene ω-amino acids Boc-3,4'-AMPB 1a and Boc-4,4'-AMPB 2a may result in an in-depth analysis of the redox-stability of these building blocks under the reducing conditions of thiol-based ligation methods. Generally, native chemical ligation allows the coupling of two unprotected peptides in neutral aqueous solution: a C-terminal thioester peptide and either an N-terminal cysteine peptide or Nα-auxiliary-capped peptides. However, for elucidating the complex redox chemistry of the two azobenzene building blocks under the reducing conditions of ligation methods, we solely applied the Boc-protected azobenzene ω-amino acid thioesters 1b and 2b instead of a C-terminal thioester peptide. We explored the conventional cysteine-based NCL with Cys-peptide 3, and also screened the application of the TFA-cleavable 4,5,6-trimethoxy-2-mercaptobenzyl (Tmb) and 1-(2,4-dimethoxyphenyl)-2-mercaptoethyl auxiliaries by using peptides 4 and 5 (Figure 1) [6-8], in order to circumvent the need for the presence of a cysteine at the ligation site.
![[1860-5397-8-101-1]](/bjoc/content/figures/1860-5397-8-101-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of azobenzene thioesters, Nα-ligation auxiliaries and peptides for the application in ligation studies. AMPB = (aminomethylphenylazo)benzoic acid.
Figure 1: Structures of azobenzene thioesters, Nα-ligation auxiliaries and peptides for the application in li...
The peptide motif in peptides 3, 4 and 5 is based on a specific binding sequence for PDZ recognition [9]. Numerous scaffolding proteins contain multiple PDZ domains for their interaction with PDZ-binding motifs at the C-terminus of transmembrane channels and receptors or other intracellular signaling proteins. Class 1 interactions involve a (S/T)-X-(V/I/L)-COOH sequence motif, and examples for these interactions include the proteins PSD-95, Fas or NHERF 1 [10]. Fas (APO-1/CD95), for example, is a cell surface receptor, belonging to the tumor-necrosis-factor receptor superfamily, which induces apoptosis. Fas-associated phosphatase-1 (FAP-1) is a Fas binding protein, which interacts with 12 to 15 of the C-terminal amino acids of the Fas receptor; however, the necessary and sufficient region for binding consists of the three C-terminal amino acids (SLV) [11-13]. PDZ-domain-containing proteins are superb examples of allosteric systems built up by semirigid domains able to interact by means of flexible regions, and therefore they seem to be ideally suited for biophysical function studies with photoswitchable ligands. For instance, Fas-associated studies in certain cells using the tripeptide SLV suggest that this small peptide alone can induce apoptosis [14,15]. Related peptides containing class I C-terminal sequence motifs, e.g., SKV, are also derived from viral origins [16]. The latter specific binding motif is associated with H5N1 influenza infections. We reasoned that the attachment of a photoswitch next to SXV PDZ-binding motifs would be a powerful strategy for exploring the function of these PDZ-binding peptide ligands in vitro and in vivo. For the synthetic ligation studies presented herein we had to place the ligation junction next to the third amino acid serine of the SKV binding motif. In the case of the conventional NCL, we used cysteine at the ligation site, and for the auxiliary mediated ligations, we decided to introduce auxiliary-glycine-conjugates at the N-terminus, due to the steric limitations of this type of ligation reaction [6-8]. After ligation, the SKV binding motif in the photoswitchable azopeptides should only be accessible in the dark trans-azo state (Scheme 1), whereas light switching to the cis-azo state should result in unfavorable interactions between the N-terminus, which follows behind the azo-switch, and the side chains that are critical for PDZ recognition (as exemplified in Scheme 1 for the 4,4'-substituted azobenzene system). However, the design presented herein, in combination with an appropriate ligation method, seems also to be applicable to the synthesis and evaluation of photoswitchable protein analogues, for example receptors with the photoswitch placed in the direct neighborhood of the specific C-terminal binding motif.
![[1860-5397-8-101-i1]](/bjoc/content/inline/1860-5397-8-101-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structural differences between the trans- and the cis-state of azopeptides with a SKV PDZ binding motif.
Scheme 1: Structural differences between the trans- and the cis-state of azopeptides with a SKV PDZ binding m...
Results and Discussion
The azobenzene ω-amino-acid thioesters 1b and 2b were prepared by following and applying literature procedures (Supporting Information File 1) [4,17]. The syntheses of the auxiliary-linked glycine conjugates 7 and 8 were accomplished by using literature protocols developed by Dawson, Offer and MacMillan [7,8] (Figure 2).
![[1860-5397-8-101-2]](/bjoc/content/figures/1860-5397-8-101-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of the glycine-linked auxiliary conjugates 7 and 8.
Figure 2: Structure of the glycine-linked auxiliary conjugates 7 and 8.
The Cys-peptide 3 and the Nα-auxiliary peptides 4 and 5 were assembled on Wang resin by manual Fmoc-based peptide synthesis using standard amino-acid building blocks (Scheme 2) [4]. Commercially available preloaded Wang resin was used, and HBTU was applied as the coupling reagent in the presence of DIPEA in NMP. Removal of the temporary Fmoc-protecting group was achieved by using 20% piperidine in NMP. Cleavage from the resin and removal of the permanent tert-butyl and trityl protecting groups was carried out with a mixture of trifluoroacetic acid, water and triisopropylsilane. The peptides 3 and 4 were purified by preparative RP-HPLC and isolated in 86% and 56% yield, respectively. Peptide 9 with a protected thiol moiety was treated with Hg(OAc)2 in TFA, followed by DTT according to the literature [8], and the deprotected peptide 5 was purified by preparative RP-HPLC and isolated in 64% yield.
![[1860-5397-8-101-i2]](/bjoc/content/inline/1860-5397-8-101-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Solid-phase synthesis of the ligation-mediating peptides 3–5.
Scheme 2: Solid-phase synthesis of the ligation-mediating peptides 3–5.
All attempts to ligate azobenzene ω-amino acid thioester 1b in phosphate buffer in the presence of TCEP·HCl and guanidinium hydrochloride failed, because of the poor solubility of the azobenzene thioester in the aqueous buffer as well as in buffer/NMP mixtures. Therefore, a ligation protocol of Danishefsky et al. was applied using DMF as the solvent [18]. The ligations were realized by reaction of the appropriate peptide 3, 4 or 5 with an excess of the azobenzene thioester (1.9 equiv), and TCEP·HCl (2.75 equiv) in the presence of Na2HPO4 (5.5 equiv, pH 7.3) at room temperature, and the reaction courses were monitored by analytical RP-HPLC (Supporting Information File 1). The ligation reactions were conducted under normal lighting conditions, and therefore small amounts of the cis-azobenzene forms of the thioesters 1b/2b and of the ligation products 10–15 were detected during HPLC monitoring (Supporting Information File 1). The results of all ligation courses prior to purification by chromatography are summarized in Table 1.
Table 1: Cysteine- and auxiliary-based ligation courses with 3,4'-AMPB thioester 1b and 4,4'-AMPB thioester 2b.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-101-i5.svg?max-width=637&scale=1.0)
|
||||||||
| No. | Thioester | Peptide | Red-1b/2bb [%] | Product | R1 | R2 | Ratio of product/red-productc | Yieldd [%] |
|---|---|---|---|---|---|---|---|---|
| 1 | 1b | 3 | – | 10 | H | CH2SH | 69:31 | 62 |
| 2 | 1b | 4 | 9 | 11 | Aux1 | H | 97:3 | 43 |
| 3 | 1b | 5 | – | 12 | Aux2 | H | 100:0 | 38 |
| 4 | 2b | 3 | 4 | 13 | H | CH2SH | 52:48 | 44 |
| 5 | 2b | 4 | 14 (12)e | 14 | Aux1 | H | 18:82 (21:79)e | 43 |
| 6 | 2b | 5 | 2 | 15 | Aux2 | H | 90:10 | 45 |
aReaction conditions: 1.9 equiv of azobenzene thioester 1b or 2b, TCEP·HCl (2.75 equiv), Na2HPO4 (5.5 equiv), DMF, rt. Peptides 3–5 were employed as the symmetric disulfides and were reduced in situ by TCEP. bConversion of 1b or 2b to the hydrazines Red-1b or Red-2b related to the unreacted excess of 1b or 2b in the ligation mixture during HPLC-monitoring after the appropriate reaction time; – : Red-1b was not detected. cHPLC-based ratio determined by HPLC-monitoring after work-up and lyophilization of the crude ligation mixture. dIsolated yield after preparative RP-HPLC. eReinvestigated ratio given in brackets.
In ligation reactions employing the N-terminal Cys-peptide 3, nearly complete conversion was determined after five hours, but stirring was nevertheless continued overnight (Table 1, entries 1 and 4). For peptide 5 containing the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary, complete consumption of the starting material was detected after 23–24 h (Table 1, entries 3 and 6). In ligation reactions employing the Nα-2-mercaptoethyl auxiliary peptide 4 reaction times were doubled for complete turnover (Table 1, entries 2 and 5). These results are in accordance with a chemoselective bimolecular thioester exchange prior to the S- to N-acyl transfer through a five-membered transition state for Cys-peptide 3, a six-membered transition state for 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary peptide 5, and a sterically more demanding five-membered transition state for racemic Nα-2-mercaptoethyl auxiliary peptide 4 [6].
During the reaction courses employing Cys-peptide 3 and the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary peptide 5, only reduction of the 4,4'-AMPB thioester 2b to the corresponding hydrazine compound Red-2b was observed, in 2–4% yield (Table 1). When using the Nα-2-mercaptoethyl auxiliary peptide 4, reduction of both azobenzene thioesters was detected: Formation of 9% Red-1b was observed for the 3,4'-AMPB thioester 1b, and of 14% Red-2b for 4,4'-AMPB thioester 2b. Obviously, the excess of the azobenzene thioester prohibits reduction of the ligation products. Additional experiments showed that the yields of the ligation products could be improved by dilution of the reaction solutions with water after complete conversion of the peptide starting materials, followed by extraction of the aqueous phase with diethylether and ethyl acetate to remove 4-acetamidothiophenol (Aatp), derived from the thioesters, as well as the excess of the azobenzene thioesters 1b and 2b. After lyophilization, analysis of the crude products by RP-HPLC indicated the formation of varying amounts of reduced ligation peptides in five out of six cases. Severe reduction was observed in ligation reactions with Cys-peptide 3 and 3,4'-AMPB thioester 1b, with a ratio of 69:31 peptide 10/peptide Red-10, as well as 4,4'-AMPB thioester 2b, with a ratio of 52:48 peptide 13/peptide Red-13 (Table 1, entries 1 and 4). Moreover, the Nα-2-mercaptoethyl auxiliary peptide 4 in combination with 4,4'-AMPB thioester 2b gave peptide 14/peptide Red14 in an 18:82 ratio (Table 1, entry 5). However, reoxidation of the hydrazine ligation peptides Red-10, Red-13 and Red-14 to the azobenzene ligation peptides 10, 13 and 14 was observed after purification by preparative RP-HPLC, and is obviously initiated by oxygen from the air. Finally, the pure ligated azobenzene peptides 10, 13 and 14 were isolated in yields of 62%, 44% and 43%, respectively. When 3,4'-AMPB thioester 1b and the Nα-2-mercaptoethyl auxiliary peptide 4 were used, the desired peptide 11 and its hydrazine analogue Red-11 were detected in a ratio of 97:3 by analytical RP-HPLC (Table 1, entry 2), and peptide 11 was isolated in 43% yield after preparative RP-HPLC. In light of the reactivity of the Nα-2-mercaptoethyl auxiliary, reduction of the azobenzene ligation peptides derived from peptide 4 can obviously only be avoided by using 3,4'-AMPB thioester 1b, due to the lower reactivity of the diazene unit in this thioester owing to the meta-substitution pattern.
The ligation reaction of 3,4'-AMPB thioester 1b with the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary peptide 5 furnished ligation peptide 12 in 38% yield (Table 1, entry 3). In this case, no hydrazine species Red-12 were detected at all (Table 1, entry 3). In addition, the ligation reaction of 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary peptide 5 and 4,4'-AMPB thioester 2b was also successfully accomplished, furnishing a ratio of 90:10 for 15/Red-15, and the pure ligation product 15 was isolated in 45% yield (Table 1, entry 6). Thus, the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary applied in peptide 5 seems to be of general use for both azobenzene systems 1b and 2b, with a manageable tendency toward reduction in the case of the 4,4'-AMPB thioester 2b.
The reduction sensitivity observed for the 4,4'-AMPB-derived thioester 2b and its ligation peptides, may be rationalized by the following considerations [19,20]: The initial addition of a thiol to an electron-poor diazene double bond is a reversible reaction, furnishing a zwitterionic adduct intermediate A en route to an N-sulfenohydrazodiarene B, with a rather labile N–S bond (Scheme 3, reaction 1). Aliphatic thiols readily undergo adduct formation towards the N-sulfenohydrazodiarene intermediates B because of their higher nucleophilicity. Accordingly, we observe a higher tendency toward reduction when applying the aliphatic Nα-2-mercaptoethyl auxiliary in comparison to the aromatic 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary. Furthermore, only 4,4'-substituted azobenzene systems, e.g., 2b and peptide 14, furnish resonance-stabilized zwitterionic intermediates A. Finally, reduction is completed by a nucleophilic attack of the second aliphatic thiol molecule at the N-sulfenohydrazodiarene B, yielding the symmetric disulfide and the respective hydrazine thioester or hydrazine peptide (Scheme 3, reaction 2). The reduction of diazene units by thiols has been intensively studied in the past for electron-poor azobenzenes or azodicarboxylates, en route to symmetrically and asymmetrically substituted disulfides [21-23].
![[1860-5397-8-101-i3]](/bjoc/content/inline/1860-5397-8-101-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reduction of the diazene unit of 4,4'-AMPB thioesters and peptides during aliphatic thiol-based Nα-auxiliary ligation strategies.
Scheme 3: Reduction of the diazene unit of 4,4'-AMPB thioesters and peptides during aliphatic thiol-based Nα-...
The final cleavage of the Boc-protecting group, the Nα-2-mercaptoethyl auxiliary and the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary in the 3,4'-AMPB ligation peptides 12 and 14 was successfully achieved with 90% TFA under standard conditions furnishing peptide 16 in 94% and 89% yield, respectively, after purification by preparative RP-HPLC (Scheme 4). Similarly, the Boc-protected 4,4'-AMPB ligation peptide 14 derived from the Nα-2-mercaptoethyl auxiliary peptide 4 was deprotected and purified yielding peptide 17 in 81% yield.
![[1860-5397-8-101-i4]](/bjoc/content/inline/1860-5397-8-101-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the azopeptides 16/17 by final TFA cleavage of the Boc-protecting groups, and of the auxiliaries Aux1 and Aux2 in the ligation peptides 11, 12 and 14.
Scheme 4: Synthesis of the azopeptides 16/17 by final TFA cleavage of the Boc-protecting groups, and of the a...
Conclusion
In conclusion, we have demonstrated the high redox stability of the Boc-protected 3,4'-AMPB thioester 1b when applying Nα-auxiliary ligation methods en route to photoswitchable peptides containing the C-terminal PDZ binding motif SKV. The positioning of the carbonyl group relative to the diazene unit in the 3,4'-AMPB-derived thioester 1b and its peptides predictably decreases the reactivity towards unwanted redox side-reactions upon application of aliphatic as well as aromatic thiols in Nα-auxiliary ligation strategies using peptides 4 and 5. In stark contrast, the Boc-protected 4,4'-AMPB thioester reproducibly furnished peptide 15 only in combination with the 4,5,6-trimethoxy-2-mercaptobenzyl auxiliary peptide 5 during the ligation course and work-up, whereas the Nα-2-mercaptoethyl auxiliary peptide 4 preferentially gave the hydrazine ligation peptide Red-14 besides the azobenzene ligation peptide 14. Our future efforts directed towards the synthesis of photoswitchable peptides will focus on water-soluble 3,4'-AMPB building blocks being applicable in native chemical ligation methods in aqueous media.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of spectra. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Wachtveitl, J.; Zumbusch, A. ChemBioChem 2011, 12, 1169–1170. doi:10.1002/cbic.201100185
Return to citation in text: [1] -
Renner, C.; Moroder, L. ChemBioChem 2006, 7, 868–878. doi:10.1002/cbic.200500531
Return to citation in text: [1] -
Beharry, A. A.; Wong, L.; Tropepe, V.; Woolley, G. A. Angew. Chem., Int. Ed. 2011, 50, 1325–1327. doi:10.1002/anie.201006506
and references cited therein.
Return to citation in text: [1] -
Rück-Braun, K.; Kempa, S.; Priewisch, B.; Richter, A.; Seedorff, S.; Wallach, L. Synthesis 2009, 4256–4267. doi:10.1055/s-0029-1217074
Return to citation in text: [1] [2] [3] -
Hoppmann, C.; Schmieder, P.; Domaing, P.; Vogelreiter, G.; Eichhorst, J.; Wiesner, B.; Morano, I.; Rück-Braun, K.; Beyermann, M. Angew. Chem., Int. Ed. 2011, 50, 7699–7702. doi:10.1002/anie.201101398
Return to citation in text: [1] -
Offer, J. Biopolymers 2010, 94, 530–541. doi:10.1002/bip.21455
Return to citation in text: [1] [2] [3] [4] -
Offer, J.; Boddy, C. N. C.; Dawson, P. E. J. Am. Chem. Soc. 2002, 124, 4642–4646. doi:10.1021/ja016731w
Return to citation in text: [1] [2] [3] -
Macmillan, D.; Anderson, D. W. Org. Lett. 2004, 6, 4659–4662. doi:10.1021/ol048145o
Return to citation in text: [1] [2] [3] [4] -
Harris, B. Z.; Lim, W. A. J. Cell Sci. 2001, 114, 3219–3231.
Return to citation in text: [1] -
Walma, T.; Spronk, C. A. E. M.; Tessari, M.; Aelen, J.; Schepens, J.; Hendriks, W.; Vuister, G. W. J. Mol. Biol. 2002, 316, 1101–1110. doi:10.1006/jmbi.2002.5402
Return to citation in text: [1] -
Yanagisawa, J.; Takahashi, M.; Kanki, H.; Yano-Yanagisawa, H.; Tazunoki, T.; Sawa, E.; Nishitoba, T.; Kamishohara, M.; Kobayashi, E.; Kataoka, S.; Sato, T. J. Biol. Chem. 1997, 272, 8539–8545. doi:10.1074/jbc.272.13.8539
Return to citation in text: [1] -
Freiss, G.; Chalbos, D. Anti-Cancer Agents Med. Chem. 2011, 11, 78–88.
Return to citation in text: [1] -
Subbaiah, V. K.; Kranjec, C.; Thomas, M.; Banks, L. Biochem. J. 2011, 439, 195–205. doi:10.1042/BJ20110903
Return to citation in text: [1] -
Ungefroren, H.; Kruse, M.-L.; Trauzold, A.; Roeschmann, S.; Roeder, C.; Arlt, A.; Henne-Bruns, D.; Kalthoff, H. J. Cell Sci. 2001, 114, 2735–2746.
Return to citation in text: [1] -
Huang, W.; Zhu, C.; Wang, H.; Horvath, E.; Eklund, E. A. J. Biol. Chem. 2008, 283, 7921–7935. doi:10.1074/jbc.M706710200
Return to citation in text: [1] -
Javier, R. T.; Rice, A. P. J. Virol. 2011, 85, 11544–11556. doi:10.1128/JVI.05410-11
Return to citation in text: [1] -
Priewisch, B.; Rück-Braun, K. J. Org. Chem. 2005, 70, 2350–2352. doi:10.1021/jo048544x
Return to citation in text: [1] -
Wu, B.; Chen, J.; Warren, J. D.; Chen, G.; Hua, Z.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2006, 45, 4116–4125. doi:10.1002/anie.200600538
Return to citation in text: [1] -
Linke, K.-H.; Brandt, W.; Göhausen, H. J. Chem. Ber. 1973, 106, 707–712. doi:10.1002/cber.19731060235
Return to citation in text: [1] -
Boulègue, C.; Löweneck, M.; Renner, C.; Moroder, L. ChemBioChem 2007, 8, 591–594. doi:10.1002/cbic.200600495
Return to citation in text: [1] -
Mukaiyama, T.; Takahashi, K. Tetrahedron Lett. 1968, 9, 5907–5908. doi:10.1016/S0040-4039(00)75437-2
Return to citation in text: [1] -
Wünsch, E.; Moroder, L.; Romani, S. Hoppe-Seyler’s Z. Physiol. Chem. 1982, 363, 1461–1464. doi:10.1515/bchm2.1982.363.2.1461
Return to citation in text: [1] -
Kosower, E. M.; Kanety-Londner, H. J. Am. Chem. Soc. 1976, 98, 3001–3007. doi:10.1021/ja00426a054
Return to citation in text: [1]
| 21. | Mukaiyama, T.; Takahashi, K. Tetrahedron Lett. 1968, 9, 5907–5908. doi:10.1016/S0040-4039(00)75437-2 |
| 22. | Wünsch, E.; Moroder, L.; Romani, S. Hoppe-Seyler’s Z. Physiol. Chem. 1982, 363, 1461–1464. doi:10.1515/bchm2.1982.363.2.1461 |
| 23. | Kosower, E. M.; Kanety-Londner, H. J. Am. Chem. Soc. 1976, 98, 3001–3007. doi:10.1021/ja00426a054 |
| 19. | Linke, K.-H.; Brandt, W.; Göhausen, H. J. Chem. Ber. 1973, 106, 707–712. doi:10.1002/cber.19731060235 |
| 20. | Boulègue, C.; Löweneck, M.; Renner, C.; Moroder, L. ChemBioChem 2007, 8, 591–594. doi:10.1002/cbic.200600495 |
| 1. | Wachtveitl, J.; Zumbusch, A. ChemBioChem 2011, 12, 1169–1170. doi:10.1002/cbic.201100185 |
| 6. | Offer, J. Biopolymers 2010, 94, 530–541. doi:10.1002/bip.21455 |
| 7. | Offer, J.; Boddy, C. N. C.; Dawson, P. E. J. Am. Chem. Soc. 2002, 124, 4642–4646. doi:10.1021/ja016731w |
| 8. | Macmillan, D.; Anderson, D. W. Org. Lett. 2004, 6, 4659–4662. doi:10.1021/ol048145o |
| 8. | Macmillan, D.; Anderson, D. W. Org. Lett. 2004, 6, 4659–4662. doi:10.1021/ol048145o |
| 18. | Wu, B.; Chen, J.; Warren, J. D.; Chen, G.; Hua, Z.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2006, 45, 4116–4125. doi:10.1002/anie.200600538 |
| 4. | Rück-Braun, K.; Kempa, S.; Priewisch, B.; Richter, A.; Seedorff, S.; Wallach, L. Synthesis 2009, 4256–4267. doi:10.1055/s-0029-1217074 |
| 5. | Hoppmann, C.; Schmieder, P.; Domaing, P.; Vogelreiter, G.; Eichhorst, J.; Wiesner, B.; Morano, I.; Rück-Braun, K.; Beyermann, M. Angew. Chem., Int. Ed. 2011, 50, 7699–7702. doi:10.1002/anie.201101398 |
| 7. | Offer, J.; Boddy, C. N. C.; Dawson, P. E. J. Am. Chem. Soc. 2002, 124, 4642–4646. doi:10.1021/ja016731w |
| 8. | Macmillan, D.; Anderson, D. W. Org. Lett. 2004, 6, 4659–4662. doi:10.1021/ol048145o |
| 2. | Renner, C.; Moroder, L. ChemBioChem 2006, 7, 868–878. doi:10.1002/cbic.200500531 |
| 3. |
Beharry, A. A.; Wong, L.; Tropepe, V.; Woolley, G. A. Angew. Chem., Int. Ed. 2011, 50, 1325–1327. doi:10.1002/anie.201006506
and references cited therein. |
| 4. | Rück-Braun, K.; Kempa, S.; Priewisch, B.; Richter, A.; Seedorff, S.; Wallach, L. Synthesis 2009, 4256–4267. doi:10.1055/s-0029-1217074 |
| 14. | Ungefroren, H.; Kruse, M.-L.; Trauzold, A.; Roeschmann, S.; Roeder, C.; Arlt, A.; Henne-Bruns, D.; Kalthoff, H. J. Cell Sci. 2001, 114, 2735–2746. |
| 15. | Huang, W.; Zhu, C.; Wang, H.; Horvath, E.; Eklund, E. A. J. Biol. Chem. 2008, 283, 7921–7935. doi:10.1074/jbc.M706710200 |
| 6. | Offer, J. Biopolymers 2010, 94, 530–541. doi:10.1002/bip.21455 |
| 7. | Offer, J.; Boddy, C. N. C.; Dawson, P. E. J. Am. Chem. Soc. 2002, 124, 4642–4646. doi:10.1021/ja016731w |
| 8. | Macmillan, D.; Anderson, D. W. Org. Lett. 2004, 6, 4659–4662. doi:10.1021/ol048145o |
| 11. | Yanagisawa, J.; Takahashi, M.; Kanki, H.; Yano-Yanagisawa, H.; Tazunoki, T.; Sawa, E.; Nishitoba, T.; Kamishohara, M.; Kobayashi, E.; Kataoka, S.; Sato, T. J. Biol. Chem. 1997, 272, 8539–8545. doi:10.1074/jbc.272.13.8539 |
| 12. | Freiss, G.; Chalbos, D. Anti-Cancer Agents Med. Chem. 2011, 11, 78–88. |
| 13. | Subbaiah, V. K.; Kranjec, C.; Thomas, M.; Banks, L. Biochem. J. 2011, 439, 195–205. doi:10.1042/BJ20110903 |
| 4. | Rück-Braun, K.; Kempa, S.; Priewisch, B.; Richter, A.; Seedorff, S.; Wallach, L. Synthesis 2009, 4256–4267. doi:10.1055/s-0029-1217074 |
| 17. | Priewisch, B.; Rück-Braun, K. J. Org. Chem. 2005, 70, 2350–2352. doi:10.1021/jo048544x |
| 10. | Walma, T.; Spronk, C. A. E. M.; Tessari, M.; Aelen, J.; Schepens, J.; Hendriks, W.; Vuister, G. W. J. Mol. Biol. 2002, 316, 1101–1110. doi:10.1006/jmbi.2002.5402 |
| 16. | Javier, R. T.; Rice, A. P. J. Virol. 2011, 85, 11544–11556. doi:10.1128/JVI.05410-11 |
© 2012 Zeyat and Rück-Braun; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)