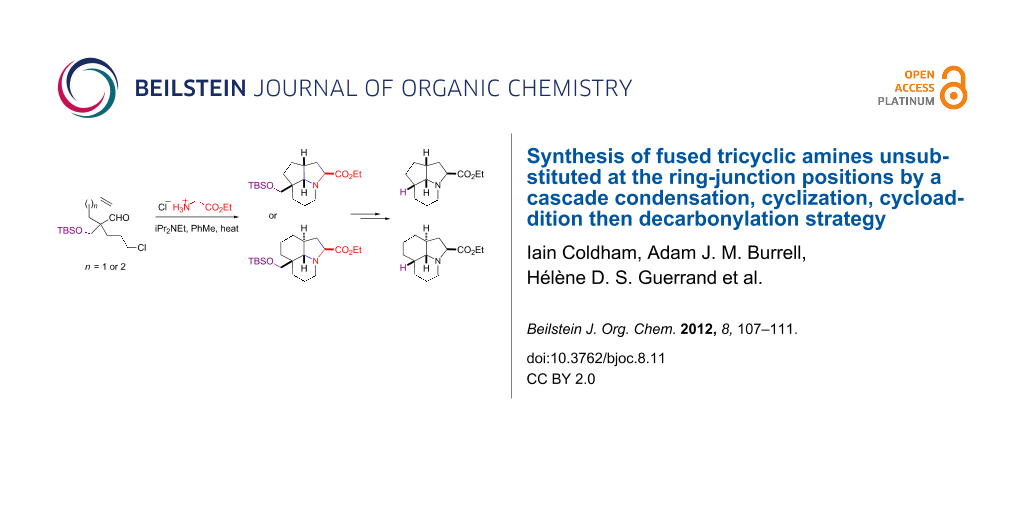
Abstract
Heating aldehydes that contain a protected hydroxymethyl group, a tethered alkyl chloride and a tethered alkenyl group at the α-position of the aldehyde with an amine sets up a cascade (tandem) reaction sequence involving condensation to an intermediate imine, then cyclization and formation of an intermediate azomethine ylide and then intramolecular dipolar cycloaddition. The fused tricyclic products are formed with complete or very high stereochemical control. The hydroxymethyl group was converted into an aldehyde – which could be removed to give the tricyclic amine products that are unsubstituted at the ring junction positions – or was converted into an alkene, which allowed the formation of the core ring system of the alkaloids scandine and meloscine.

Graphical Abstract
Introduction
Cascade reaction sequences [1] provide a rapid and efficient means to build complexity in organic chemistry. One such sequence involves a cyclization followed by in situ intramolecular cycloaddition to give three new rings in a single transformation [2-8]. We have been studying the intramolecular dipolar cycloaddition of azomethine ylides in synthesis [9-16] and were able to show that the azomethine ylide could be prepared in situ by a cyclization step [17,18]; for example, by heating the aldehyde 1 (R = Et) with glycine ethyl ester to give the tricyclic product 2 [19,20] (Scheme 1). In this chemistry, the intermediate imine undergoes cyclization onto the tethered alkyl chloride to give an iminium ion which deprotonates to give an azomethine ylide that undergoes dipolar cycloaddition with the alkene. A single stereoisomer of product 2 was formed. Alternatively, glycine could be used and led to a tricyclic product via an intermediate azomethine ylide formed by decarboxylation. Unfortunately, when the substrate 1 (R = H) was used, heating with glycine or glycine ethyl ester did not yield the desired tricyclic products due to competing enamine formation [21].
![[1860-5397-8-11-i1]](/bjoc/content/inline/1860-5397-8-11-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cascade chemistry for 2-ethyl-aldehydes.
Scheme 1: Cascade chemistry for 2-ethyl-aldehydes.
For this cascade cyclization–cycloaddition chemistry to be widely applicable (for example for the preparation of alkaloids), we need to be able to access tricyclic products with no substituents (only hydrogen atoms) at the ring junction positions (such as compound 2, R = H, or other ring sizes) [22]. This paper outlines an approach to such targets using a substituent (to block side reactions) which can then be removed by decarbonylation [23].
Results and Discussion
Preparation of the aldehyde substrates
We decided to incorporate a one-carbon substituent, alpha to the aldehyde group, that could potentially be removed after the cascade cyclization–cycloaddition chemistry. Starting from nitrile 3, we prepared three different aldehydes, as shown in Scheme 2 and Scheme 3. Double deprotonation (of the alcohol and alpha to the nitrile) of substrate 3 and C-alkylation occurred smoothly to give 4a or 4b (n = 1 or 2, respectively). A second double deprotonation and then an alkylation with bromochloropropane gave 5a and 5b. Oxidation of the alcohol 5a (n = 1) gave the aldehyde 6. Alternatively, protection of 5a and 5b as their tert-butyldimethylsilyl ethers gave 7a and 7b. DIBAL-H reduction of nitrile 7b (n = 2) gave aldehyde 8b. The same reduction of nitrile 7a gave a low yield of the desired aldehyde 8a; so, an alternative strategy to prepare the aldehyde with one less methylene group in the side-chain was studied (Scheme 3). Alkylation of the commercially available substituted malonate 9 gave product 10 together with diethyl cyclobutanedicarboxylate (formed by cyclization of the malonate anion onto the internal alkyl chloride). This by-product was removed by column chromatography after reduction of both esters to give diol 11. Mono-protection gave the alcohol 12 which was oxidized to aldehyde 8a.
![[1860-5397-8-11-i2]](/bjoc/content/inline/1860-5397-8-11-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of aldehydes 6 and 8b. Step a: LDA, THF, 0 °C, CH2=CHCH2(CH2)nBr (n = 1, 56%; n = 2, 76%). Step b: LDA, THF, −78 °C, ClCH2CH2CH2Br (n = 1, 80%; n = 2, 89%). Step c: Using 5a, (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to rt, 92%. Step d: TBSCl, imidazole, CH2Cl2, rt, 16 h (n = 1, 92%; n = 2, 94%). Step e: Using 7b, DIBAL-H, CH2Cl2, −78 °C, 2 h then HCl(aq) (2 M), 62%.
Scheme 2: Preparation of aldehydes 6 and 8b. Step a: LDA, THF, 0 °C, CH2=CHCH2(CH2)nBr (n = 1, 56%; n = 2, 76...
![[1860-5397-8-11-i3]](/bjoc/content/inline/1860-5397-8-11-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of aldehyde 8a. Step a: NaH, THF, 0 to 70 °C, CH2=CHCH2CH2Br. Step b: DIBAL-H, CH2Cl2, 0 °C, 2 h then HCl(aq) (2 M), 24% over two steps. Step c: TBSCl, imidazole, CH2Cl2, rt, 16 h, 86%. Step d: (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to rt, 96%.
Scheme 3: Preparation of aldehyde 8a. Step a: NaH, THF, 0 to 70 °C, CH2=CHCH2CH2Br. Step b: DIBAL-H, CH2Cl2, ...
Cascade condensation, cyclization, cycloaddition
The cascade tricyclic ring formation was then investigated with the aldehydes 6, 8a and 8b. Heating aldehyde 6 with glycine gave a mixture of unidentifiable products. However, heating with glycine ethyl ester in toluene gave the desired tricyclic product 13a as the major stereoisomer, together with a small amount of the separable stereosiomer 13b (Scheme 4). The major isomer 13a was assumed to have the all-cis configuration based on related chemistry (compare with product 2 [19,20], Scheme 1). The minor isomer was assigned by NOESY studies, in which there was a strong enhancement between the ring junction protons and a strong enhancement between the proton alpha to the ester and the ring junction proton alpha to the nitrogen. The reaction is supposed to proceed by initial imine formation, then cyclization with displacement of chloride to give an iminium ion. Deprotonation of this iminium ion would give the azomethine ylide that undergoes intramolecular cycloaddition. The preference for the all-cis isomer 13a arises from a preference for cycloaddition from the conformation shown in Scheme 4. Unfortunately, we were not able to remove the nitrile substituent from the product 13a using lithium in ammonia [24], which instead converted the ethyl ester to its primary carboxylic amide, or using DIBAL-H, which gave a complex mixture of products.
![[1860-5397-8-11-i4]](/bjoc/content/inline/1860-5397-8-11-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cascade chemistry with aldehyde 6 and preferred conformation of intermediate azomethine ylide during cycloaddition.
Scheme 4: Cascade chemistry with aldehyde 6 and preferred conformation of intermediate azomethine ylide durin...
Heating aldehyde 8a with glycine gave cycloadduct 14 as a single diastereoisomer (Scheme 5). Deprotection of the silyl ether with tetra-n-butylammonium fluoride (TBAF) gave the alcohol 15, then Swern oxidation yielded aldehyde 16. The stereochemistry of alcohol 15 was confirmed by single crystal X-ray analysis. This stereochemistry was expected for these cascade reactions, in which the substituent alpha to the aldehyde (in this case the TBSOCH2 group) is located in the transition state cis to the developing ring junction protons. The next step, to remove the one-carbon unit to provide the desired tricyclic product with all ring-junction protons, was a decarbonylation and this type of reaction is known using Wilkinson’s catalyst [25]. We were disappointed to find that attempts to conduct this decarbonylation using aldehyde 16 and [Rh(PPh3)3Cl] resulted only in decomposition.
![[1860-5397-8-11-i5]](/bjoc/content/inline/1860-5397-8-11-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cascade chemistry with aldehyde 8a and glycine.
Scheme 5: Cascade chemistry with aldehyde 8a and glycine.
As an aside, we were interested in using the products of this chemistry for the construction of the core ring system found in alkaloid natural products. Wittig reaction with aldehyde 16 should be possible to provide the core ring system found in the alkaloids meloscine and scandine [26-30]. However, we chose to investigate a shorter reaction sequence without the need for protecting groups and to carry out the Wittig reaction at an earlier stage, as shown in Scheme 6. Olefination of aldehyde 6 with the anion formed from methyltriphenylphosphonium bromide gave alkene 17, which was reduced with DIBAL-H to give aldehyde 18. Heating this aldehyde with glycine in toluene gave the desired tricyclic product 19 as a single stereoisomer. NOESY studies verified the stereochemistry as shown. Only one of the two alkene groups acts as the dienophile, as expected for conformational reasons. This synthesis (six steps from nitrile 3) represents an efficient entry to the core of the alkaloids meloscine and scandine, which contains the same fused tricyclic ring system.
![[1860-5397-8-11-i6]](/bjoc/content/inline/1860-5397-8-11-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of the core tricyclic ring system of meloscine and scandine.
Scheme 6: Synthesis of the core tricyclic ring system of meloscine and scandine.
The use of glycine to form a ‘non-stabilized’ ylide had led to the desired cycloadduct 14, although subsequent decarbonylation had not been successful using aldehyde 16 (Scheme 5). Despite this, we chose to investigate the cascade chemistry using glycine ethyl ester (to form a ‘stabilized’ ylide), followed by possible later decarbonylation.
Heating aldehydes 8a or 8b with glycine ethyl ester in toluene gave the expected tricyclic products (Scheme 7). Using aldehyde 8a, a single stereoisomer was formed (assumed to have the all-cis configuration shown and confirmed by NOESY studies). Using aldehyde 8b, the major product was the stereoisomer 21, which was expected based on related chemistry [19] and verified by the large coupling constant in the 1H NMR spectrum for the ring junction proton alpha to the nitrogen atom (2.20 ppm, doublet, J = 11.5 Hz). However, this transformation was not entirely stereoselective and a (separable) mixture of two other stereoisomers 22 and 23 was also formed (NOESY studies were used to ascertain the relative stereochemistry).
![[1860-5397-8-11-i7]](/bjoc/content/inline/1860-5397-8-11-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Cascade chemistry with aldehydes 8a and 8b and glycine ethyl ester.
Scheme 7: Cascade chemistry with aldehydes 8a and 8b and glycine ethyl ester.
To complete the desired aim to provide a method to prepare fused tricyclic products with all ring-junction protons, we carried out the transformations shown in Scheme 8. Desilylation of 20 and 21 gave the alcohols 24 and 27, respectively. Swern oxidation then gave the aldehydes 25 and 28. Finally, using Wilkinson’s catalyst, we were pleased to find that both these aldehyde substrates were amenable to decarbonylation. The optimum conditions were found to be a temperature of about 150 °C for about 2 h. The decarbonylation occurred with overall retention of stereochemistry, as verified by NOESY studies. The presence of the ethyl ester substituent must be sufficient to allow a successful decarbonylation (in comparison with substrate 16), possibly due to the reduced ability of the amine nitrogen atom to coordinate to the rhodium complex.
![[1860-5397-8-11-i8]](/bjoc/content/inline/1860-5397-8-11-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Decarbonylation reactions to give the products 26 and 29.
Scheme 8: Decarbonylation reactions to give the products 26 and 29.
Conclusion
The chemistry described here provides a method to prepare fused tricyclic products in which the ring junction positions are unsubstituted. Problems with conducting the cascade condensation, cyclisation, cycloaddition chemistry using azomethine ylides derived from enolisable aldehyde substrates [21] has been circumvented by incorporating a one-carbon unit at the alpha-position of the aldehyde. This one-carbon unit can be removed in certain cases using a decarbonylation reaction.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectroscopic data. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Nicolaou, K. C.; Chen, J. S. Chem. Soc. Rev. 2009, 38, 2993. doi:10.1039/b903290h
Return to citation in text: [1] -
Brasholz, M.; Macdonald, J. M.; Saubern, S.; Ryan, J. H.; Holmes, A. B. Chem.–Eur. J. 2010, 16, 11471. doi:10.1002/chem.201001435
Return to citation in text: [1] -
Brasholz, M.; Johnson, B. A.; Macdonald, J. M.; Polyzos, A.; Tsanaktsidis, J.; Saubern, S.; Holmes, A. B.; Ryan, J. H. Tetrahedron 2010, 66, 6445. doi:10.1016/j.tet.2010.04.092
Return to citation in text: [1] -
Flick, A. C.; Caballero, M. J. A.; Padwa, A. Tetrahedron 2010, 66, 3643. doi:10.1016/j.tet.2010.03.088
Return to citation in text: [1] -
Flick, A. C.; Caballero, M. J. A.; Lee, H. I.; Padwa, A. J. Org. Chem. 2010, 75, 1992. doi:10.1021/jo100055u
Return to citation in text: [1] -
Stearman, C. J.; Wilson, M.; Padwa, A. J. Org. Chem. 2009, 74, 3491. doi:10.1021/jo9003579
Return to citation in text: [1] -
Karatholuvhu, M. S.; Sinclair, A.; Newton, A. F.; Alcaraz, M.-L.; Stockman, R. A.; Fuchs, P. L. J. Am. Chem. Soc. 2006, 128, 12656. doi:10.1021/ja065015x
Return to citation in text: [1] -
Padwa, A. Chem. Soc. Rev. 2009, 38, 3072. doi:10.1039/b816701j
Return to citation in text: [1] -
Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765. doi:10.1021/cr040004c
Return to citation in text: [1] -
Burrell, A. J. M.; Coldham, I. Curr. Org. Synth. 2010, 7, 312. doi:10.2174/157017910791414472
Return to citation in text: [1] -
Coldham, I.; Coles, S. J.; Crapnell, K. M.; Fernàndez, J.-C.; Haxell, T. F. N.; Hursthouse, M. B.; Moseley, J. D.; Treacy, A. B. Chem. Commun. 1999, 1757. doi:10.1039/a904667d
Return to citation in text: [1] -
Coldham, I.; Crapnell, K. M.; Fernàndez, J.-C.; Moseley, J. D.; Rabot, R. J. Org. Chem. 2002, 67, 6181. doi:10.1021/jo016376s
Return to citation in text: [1] -
Coldham, I.; Pih, S. M.; Rabot, R. Synlett 2005, 1743. doi:10.1055/s-2005-871538
Return to citation in text: [1] -
Coldham, I.; Dobson, B. C.; Franklin, A. I.; Fletcher, S. R. Tetrahedron Lett. 2007, 48, 873. doi:10.1016/j.tetlet.2006.11.130
Return to citation in text: [1] -
Coldham, I.; Dobson, B. C.; Fletcher, S. R.; Franklin, A. I. Eur. J. Org. Chem. 2007, 2676. doi:10.1002/ejoc.200700045
Return to citation in text: [1] -
Franklin, A. I.; Bensa, D.; Adams, H.; Coldham, I. Org. Biomol. Chem. 2011, 9, 1901. doi:10.1039/c0ob01019g
Return to citation in text: [1] -
Pearson, W. H.; Stoy, P.; Mi, Y. J. Org. Chem. 2004, 69, 1919. doi:10.1021/jo030334h
Return to citation in text: [1] -
Pearson, W. H.; Kropf, J. E.; Choy, A. L.; Lee, I. Y.; Kampf, J. W. J. Org. Chem. 2007, 72, 4135. doi:10.1021/jo0703799
Return to citation in text: [1] -
Coldham, I.; Burrell, A. J. M.; White, L. E.; Adams, H.; Oram, N. Angew. Chem., Int. Ed. 2007, 46, 6159. doi:10.1002/anie.200701943
Return to citation in text: [1] [2] [3] -
Burrell, A. J. M.; Coldham, I.; Watson, L.; Oram, N.; Pilgram, C. D.; Martin, N. G. J. Org. Chem. 2009, 74, 2290. doi:10.1021/jo8019913
Return to citation in text: [1] [2] -
Burrell, A. J. M.; Coldham, I.; Oram, N. Org. Lett. 2009, 11, 1515. doi:10.1021/ol9001653
Return to citation in text: [1] [2] -
Burrell, A. J. M.; Watson, L.; Martin, N. G.; Oram, N.; Coldham, I. Org. Biomol. Chem. 2010, 8, 4530. doi:10.1039/c0ob00408a
Return to citation in text: [1] -
Coldham, I.; Burrell, A. J. M.; Watson, L.; Oram, N.; Martin, N. G. Heterocycles 2012, 84, 597. doi:10.3987/COM-11-S(P)29
(Example for a desulfurization).
Return to citation in text: [1] -
Vilsmaier, E.; Milch, G.; Bergsträßer, T. Tetrahedron 1998, 54, 6403. doi:10.1016/S0040-4020(98)00298-1
Return to citation in text: [1] -
Tsuji, J.; Ohno, K. Synthesis 1969, 157. doi:10.1055/s-1969-34197
Return to citation in text: [1] -
Hugel, G.; Levy, J. J. Org. Chem. 1986, 51, 1594. doi:10.1021/jo00359a040
Return to citation in text: [1] -
Zhang, H.; Curran, D. P. J. Am. Chem. Soc. 2011, 133, 10376. doi:10.1021/ja2042854
Return to citation in text: [1] -
Hayashi, Y.; Inagaki, F.; Mukai, C. Org. Lett. 2011, 13, 1778. doi:10.1021/ol200311y
Return to citation in text: [1] -
Selig, P.; Herdtweck, E.; Bach, T. Chem.–Eur. J. 2009, 15, 3509. doi:10.1002/chem.200802383
Return to citation in text: [1] -
Overman, L. E.; Robertson, G. M.; Robichaud, A. J. J. Am. Chem. Soc. 1991, 113, 2598. doi:10.1021/ja00007a038
Return to citation in text: [1]
| 1. | Nicolaou, K. C.; Chen, J. S. Chem. Soc. Rev. 2009, 38, 2993. doi:10.1039/b903290h |
| 19. | Coldham, I.; Burrell, A. J. M.; White, L. E.; Adams, H.; Oram, N. Angew. Chem., Int. Ed. 2007, 46, 6159. doi:10.1002/anie.200701943 |
| 20. | Burrell, A. J. M.; Coldham, I.; Watson, L.; Oram, N.; Pilgram, C. D.; Martin, N. G. J. Org. Chem. 2009, 74, 2290. doi:10.1021/jo8019913 |
| 17. | Pearson, W. H.; Stoy, P.; Mi, Y. J. Org. Chem. 2004, 69, 1919. doi:10.1021/jo030334h |
| 18. | Pearson, W. H.; Kropf, J. E.; Choy, A. L.; Lee, I. Y.; Kampf, J. W. J. Org. Chem. 2007, 72, 4135. doi:10.1021/jo0703799 |
| 9. | Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765. doi:10.1021/cr040004c |
| 10. | Burrell, A. J. M.; Coldham, I. Curr. Org. Synth. 2010, 7, 312. doi:10.2174/157017910791414472 |
| 11. | Coldham, I.; Coles, S. J.; Crapnell, K. M.; Fernàndez, J.-C.; Haxell, T. F. N.; Hursthouse, M. B.; Moseley, J. D.; Treacy, A. B. Chem. Commun. 1999, 1757. doi:10.1039/a904667d |
| 12. | Coldham, I.; Crapnell, K. M.; Fernàndez, J.-C.; Moseley, J. D.; Rabot, R. J. Org. Chem. 2002, 67, 6181. doi:10.1021/jo016376s |
| 13. | Coldham, I.; Pih, S. M.; Rabot, R. Synlett 2005, 1743. doi:10.1055/s-2005-871538 |
| 14. | Coldham, I.; Dobson, B. C.; Franklin, A. I.; Fletcher, S. R. Tetrahedron Lett. 2007, 48, 873. doi:10.1016/j.tetlet.2006.11.130 |
| 15. | Coldham, I.; Dobson, B. C.; Fletcher, S. R.; Franklin, A. I. Eur. J. Org. Chem. 2007, 2676. doi:10.1002/ejoc.200700045 |
| 16. | Franklin, A. I.; Bensa, D.; Adams, H.; Coldham, I. Org. Biomol. Chem. 2011, 9, 1901. doi:10.1039/c0ob01019g |
| 19. | Coldham, I.; Burrell, A. J. M.; White, L. E.; Adams, H.; Oram, N. Angew. Chem., Int. Ed. 2007, 46, 6159. doi:10.1002/anie.200701943 |
| 2. | Brasholz, M.; Macdonald, J. M.; Saubern, S.; Ryan, J. H.; Holmes, A. B. Chem.–Eur. J. 2010, 16, 11471. doi:10.1002/chem.201001435 |
| 3. | Brasholz, M.; Johnson, B. A.; Macdonald, J. M.; Polyzos, A.; Tsanaktsidis, J.; Saubern, S.; Holmes, A. B.; Ryan, J. H. Tetrahedron 2010, 66, 6445. doi:10.1016/j.tet.2010.04.092 |
| 4. | Flick, A. C.; Caballero, M. J. A.; Padwa, A. Tetrahedron 2010, 66, 3643. doi:10.1016/j.tet.2010.03.088 |
| 5. | Flick, A. C.; Caballero, M. J. A.; Lee, H. I.; Padwa, A. J. Org. Chem. 2010, 75, 1992. doi:10.1021/jo100055u |
| 6. | Stearman, C. J.; Wilson, M.; Padwa, A. J. Org. Chem. 2009, 74, 3491. doi:10.1021/jo9003579 |
| 7. | Karatholuvhu, M. S.; Sinclair, A.; Newton, A. F.; Alcaraz, M.-L.; Stockman, R. A.; Fuchs, P. L. J. Am. Chem. Soc. 2006, 128, 12656. doi:10.1021/ja065015x |
| 8. | Padwa, A. Chem. Soc. Rev. 2009, 38, 3072. doi:10.1039/b816701j |
| 21. | Burrell, A. J. M.; Coldham, I.; Oram, N. Org. Lett. 2009, 11, 1515. doi:10.1021/ol9001653 |
| 19. | Coldham, I.; Burrell, A. J. M.; White, L. E.; Adams, H.; Oram, N. Angew. Chem., Int. Ed. 2007, 46, 6159. doi:10.1002/anie.200701943 |
| 20. | Burrell, A. J. M.; Coldham, I.; Watson, L.; Oram, N.; Pilgram, C. D.; Martin, N. G. J. Org. Chem. 2009, 74, 2290. doi:10.1021/jo8019913 |
| 23. |
Coldham, I.; Burrell, A. J. M.; Watson, L.; Oram, N.; Martin, N. G. Heterocycles 2012, 84, 597. doi:10.3987/COM-11-S(P)29
(Example for a desulfurization). |
| 26. | Hugel, G.; Levy, J. J. Org. Chem. 1986, 51, 1594. doi:10.1021/jo00359a040 |
| 27. | Zhang, H.; Curran, D. P. J. Am. Chem. Soc. 2011, 133, 10376. doi:10.1021/ja2042854 |
| 28. | Hayashi, Y.; Inagaki, F.; Mukai, C. Org. Lett. 2011, 13, 1778. doi:10.1021/ol200311y |
| 29. | Selig, P.; Herdtweck, E.; Bach, T. Chem.–Eur. J. 2009, 15, 3509. doi:10.1002/chem.200802383 |
| 30. | Overman, L. E.; Robertson, G. M.; Robichaud, A. J. J. Am. Chem. Soc. 1991, 113, 2598. doi:10.1021/ja00007a038 |
| 22. | Burrell, A. J. M.; Watson, L.; Martin, N. G.; Oram, N.; Coldham, I. Org. Biomol. Chem. 2010, 8, 4530. doi:10.1039/c0ob00408a |
| 21. | Burrell, A. J. M.; Coldham, I.; Oram, N. Org. Lett. 2009, 11, 1515. doi:10.1021/ol9001653 |
| 24. | Vilsmaier, E.; Milch, G.; Bergsträßer, T. Tetrahedron 1998, 54, 6403. doi:10.1016/S0040-4020(98)00298-1 |
© 2012 Coldham et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)