Abstract
Three new isomeric asymmetric diarylethenes with a naphthyl moiety and a formyl group at the para, meta or ortho position of the terminal benzene ring were synthesized. Their photochromism, fluorescent-switch, and electrochemical properties were investigated. Among these diarylethenes, the one with a formyl group at the ortho position of benzene displayed the largest molar absorption coefficients and fluorescence quantum yield. The cyclization quantum yields of these compounds increased in the order of para < ortho < meta, whereas their cycloreversion quantum yields decreased in the order of meta > para > ortho. Additionally, all of these diarylethenes functioned as effective fluorescent switches in both solution and PMMA films. Cyclic voltammograms proved that the formyl group and its position could effectively modulate the electrochemical behaviors of these diarylethene derivatives.

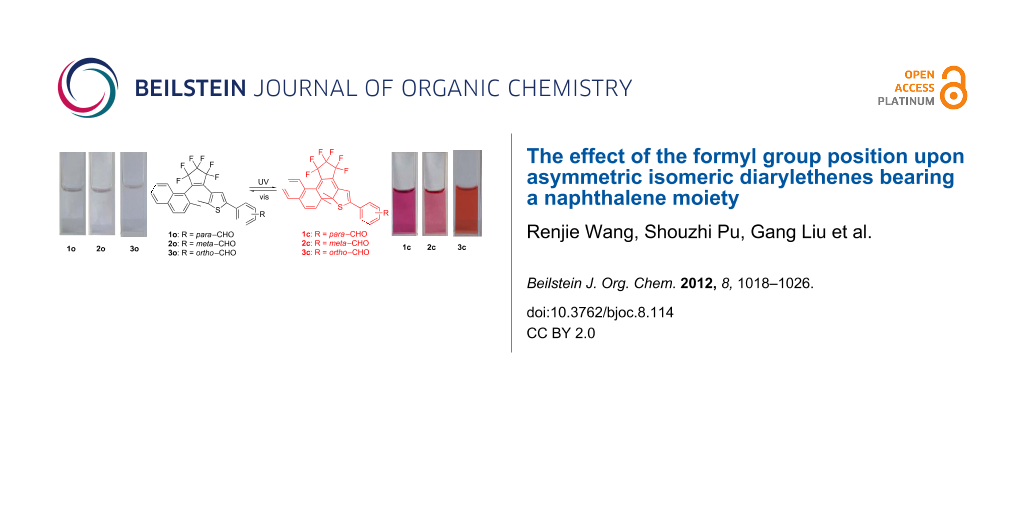
Graphical Abstract
Introduction
In the past decade, photochromic materials have received much attention because of their applications in potential photoswitchable, molecular devices and optical memory storage systems [1]. Among these materials, the utilization of diarylethene derivatives in molecular electronics, optical memory, and variable-transmission filters has been well documented [1-3]. So far, a huge number of studies concerning the photochromic properties of dithienylethene derivatives have been reported [4-14].
Light is a convenient and powerful trigger to control the reactivity of biomolecules in organisms, exemplified by its use in fluorescent probes, fluorescence imaging, and molecular switches in logic circuits [15-22]. In general, in order to efficiently realize the artificial induction of photosensitivity in biomolecules, the photoactive molecules must possess the following properties: (1) low cytotoxicity, (2) high sensitivity, (3) easy chemical modification. In the past few decades, various types of photochromic molecules, such as fulgides, spiropyranes, azobenzenes, and diarylethenes, have been developed [2,23-31].
Among these compounds, diarylethene is one of the most promising photoswitchable units within the photochromic system, and the successful use of diarylethenes as a fluorescence modulation center to realize a photoswitchable probe for imaging living cells was reported. For example, Zou et al. reported an amphiphilic molecule with hydrophilic and hydrophobic chains on two ends of a rigid diarylethene core. This compound can form stable vesicle nanostructures in aqueous solution, and exhibits excellent switchable fluorescence between open and closed states in the living cells, with low cytotoxicity [32]. Piao et al. developed a multiresponsive fluorescent molecular switch containing terpyridine. This diarylethene can serve as a detector for metal-ion transmembrane transport [33]. Singer et al. explored a novel diarylethene with a 7-deazaadenosine, which led to new research in photochromic nucleosides and molecular recognition properties of nucleic acids with the light sensitivity of diarylethenes [34]. Recently, Wu et al. designed and synthesized a novel diarylethene-containing dithiazolethene, which exhibited a gated photochromic reactivity controlled by complexation/dissociation with BF3 [35]. All of the above research revealed that the explored novel diarylethenes have versatile applications and are still important and attractive.
Diarylethene derivatives, especially those containing a perfluorocyclopentene bridge are one of the most promising photochromic compounds due to their high fatigue resistance and thermal stability [2,36]. In general, the photochromic reactivity of diarylethenes mainly depends on heteroaryl groups and different electron-donor/acceptor substituents. The formyl group can be modified to form various chemical groups and it can also be connected with different fluorophores via a Schiff base structure. In a previous work, we developed a new class of diarylethenes with a naphthalene group and a thiophene group. The results revealed that these molecules have excellent photochromism with good fatigue resistance and thermal stability [37]. In this study, in order to further elucidate the substituent position effects on the photochromic features of naphthalene-containing diarylethenes, we synthesized three new isomeric diarylethenes with a formyl group at the para, meta, and ortho position on the terminal benzene ring (1–3). The photochromic scheme of 1–3 is shown in Scheme 1.
![[1860-5397-8-114-i1]](/bjoc/content/inline/1860-5397-8-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochromism of diarylethenes 1–3.
Scheme 1: Photochromism of diarylethenes 1–3.
Results and Discussion
The synthesis route for diarylethenes 1o–3o is shown in Scheme 2. First, the benzaldehydethiophene derivatives 5a–c were prepared by Suzuki coupling of three bromobenzaldehyde derivatives with a thiopheneboronic acid 4 [38-42]. Second, 1,3-dioxolane-phenylthiophene derivatives 6a–c were prepared by the reported method [39-41,43]. Then, 1,3-dioxolane-phenylthiophene derivatives 6a–c were separately lithiated and coupled with (2-methylnaphth-1-yl)perfluorocyclopentene [37] to give the asymmetric diarylethene derivatives 7a–c [44]. Finally, compounds 1o–3o were prepared by hydrolyzing compounds 7a–c in the presence of pyridine and p-toluenesulfonic acid in acetone/water. The structures of 1o–3o were confirmed by elemental analysis, NMR, and IR (Supporting Information File 1).
![[1860-5397-8-114-i2]](/bjoc/content/inline/1860-5397-8-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic route for diarylethenes 1–3.
Scheme 2: Synthetic route for diarylethenes 1–3.
Photoisomerization of diarylethenes 1–3
Diarylethenes 1−3 showed good photochromic properties and could be toggled between their colorless open-ring isomers (1o−3o) and colored closed-ring isomers (1c−3c) by alternate irradiation with UV and visible light (λ > 500 nm). As shown in Figure 1A, diarylethene 1o exhibited a sharp absorption peak at 323 nm (ε, 2.82 × 104 L mol−1 cm−1) in hexane, which arose from the π→π* transition [45]. Upon irradiation with 297 nm light, the colorless solution of 1o gradually turned red, and a new absorption band was observed in the visible region centered at 524 nm (ε, 1.40 × 104 L mol−1 cm−1) due to the formation of the closed-ring isomer 1c. Alternatively, the red-colored solution could be bleached to become colorless by re-production of the open-ring isomer 1o upon irradiation with visible light (λ > 500 nm). In the photostationary state, a clear isosbestic point of diarylethene 1 was observed at 349 nm, which supported the reversible two-component photochromic reaction scheme [46]. Similarly 1o, compounds 2o and 3o also showed good photochromism in hexane (Figure 1B). The colorless solutions of 2o and 3o turned pink and magenta due to the formation of the closed-ring isomers 2c and 3c, when irradiated with 297 nm light. Their absorption maxima appeared at 512 and 496 nm respectively. The colored solutions of 2c and 3c can also be decolorized upon irradiation with visible light (λ > 500 nm), and their isosbestic points were observed at 277 and 268 nm, respectively. The color changes of diarylethenes 1–3 by alternating irradiation with UV and visible light (λ > 500 nm) in hexane are shown in Figure 2A.
![[1860-5397-8-114-1]](/bjoc/content/figures/1860-5397-8-114-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Absorption spectral changes of diarylethenes 1–3 by photoirradiation with UV–vis in hexane (2.0 × 10−5 mol/L) at room temperature: (A) 1; (B) 2 and 3.
Figure 1: Absorption spectral changes of diarylethenes 1–3 by photoirradiation with UV–vis in hexane (2.0 × 10...
![[1860-5397-8-114-2]](/bjoc/content/figures/1860-5397-8-114-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The color changes of diarylethene 1–3 by photoirradiation at room temperature: (A) in hexane; (B) in PMMA films.
Figure 2: The color changes of diarylethene 1–3 by photoirradiation at room temperature: (A) in hexane; (B) i...
In PMMA amorphous films, diarylethenes 1–3 also showed similar photochromic activity to that in hexane. The absorption maxima of closed-ring isomers of diarylethenes 1c–3c in PMMA films were at longer wavelengths. The values of the absorption maxima of the ring-closed isomers are 14 nm for 1c, 7 nm for 2c, and 21 nm for 3c. The redshift phenomena may be ascribed to a polar effect of the polymer matrix and the stabilization of the molecular arrangement in the solid medium [47,48]. The color changes of diarylethenes 1–3 upon alternating irradiation with UV and visible light in PMMA films are shown in Figure 2B. The photoconversion ratios from open-ring to closed-ring isomers of 1–3 were analyzed by HPLC in the photostationary state (Figure 3). It was calculated that their photoconversion ratios in the photostationary state were 82% for 1, 79% for 2, and 81% for 3.
![[1860-5397-8-114-3]](/bjoc/content/figures/1860-5397-8-114-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The photoconversion ratios of diarylethenes 1–3 in the photostationary state as analyzed by HPLC.
Figure 3: The photoconversion ratios of diarylethenes 1–3 in the photostationary state as analyzed by HPLC.
The photochromic features of compounds 1–3 are summarized in Table 1. The results indicate that the position of the formyl group at the terminal benzene significantly affects the photochromic properties of these diarylethenes, such as the absorption maxima, molar absorption coefficients, and quantum yields of cyclization and cycloreversion. For the isomeric diarylethenes 1–3, the absorption maxima of both the open-ring and closed-ring isomers exhibited a remarkable hypochromatic shift when the formyl group was moved from the para to the meta, then to the ortho position in both hexane and PMMA films, whereas the molar absorption coefficients of diarylethenes 1–3 increased in order of meta < para < ortho substitution by the formyl group in hexane. The results were in agreement with those of the reported diarylethenes containing an electron-withdrawing cyano group [49], but were different from those with an electron-donating methoxy group [14,50,51]. The cycloreversion quantum yields of diarylethenes 1–3 increased in the order of ortho (Φc-o = 0.11) < para (Φc-o = 0.12) < meta substitution (Φc-o = 0.15) by the formyl group. However, the cyclization quantum yield of the para-substituted derivative 1 was the largest (Φc-o = 0.35), while that of the meta-substituted derivative 2 was the lowest (Φc-o = 0.21). Compared to the electron-donating methoxy group or electron-withdrawing cyano group [49,50], the position of the electron-withdrawing formyl group can effectively modulate the absorption maxima of diarylethenes, which may be a novel strategy for exploring photochromic diarylethenes at shorter wavelengths.
Table 1: Absorption spectral properties of diarylethenes 1–3 in hexane (2.0 × 10−5 mol L−1) and in PMMA films (10%, w/w) at room temperature.
| compound | λo,max/nma (ε/L mol−1 cm−1) | λc,max/nmb (ε/L mol−1 cm−1) | Φc | conversion at PSS in hexane | |||
|---|---|---|---|---|---|---|---|
| hexane | PMMA film | hexane | PMMA film | Φo-c | Φc-o | ||
| 1 | 323 (2.82 × 104) | 326 | 524 (1.40 × 104) | 538 | 0.35 | 0.12 | 82 |
| 2 | 256 (2.75 × 104) | 288 | 512 (1.01 × 104) | 519 | 0.21 | 0.15 | 79 |
| 3 | 245 (3.48 × 104) | 248 | 496 (1.57 × 104) | 517 | 0.25 | 0.11 | 81 |
aAbsorption maxima of ring-open isomers. bAbsorption maxima of ring-closed isomers. cQuantum yields of ring-open (Φo-c) and ring-closed isomers (Φc-o), respectively.
The thermal stabilities of the open-ring and closed-ring isomers of 1–3 were tested by storing the compounds at both room temperature in hexane and at 351 K in ethanol. The hexane solutions were kept at room temperature in the dark, and exposed to air for more than two months. No changes in the UV–vis spectra were observed for 1–3. At 351 K, diarylethenes 1–3 also showed excellent thermal stability for more than 12 h in ethanol. Fatigue resistance is a critical factor for practical applications in optical devices, and the fatigue resistances of diarylethenes 1–3 were examined in both hexane and PMMA films by alternate irradiation with UV and visible light at room temperature [2,52]. As shown in Figure 4, the coloration and decoloration cycle of 1–3 can be repeated more than 100 times in hexane with less than 5% degradation of 1c–3c. In PMMA films, 1–3 also exhibited excellent photochromic properties after 200 cycles with only ca. 6–10% degradation of 1c–3c. The results showed that all three isomeric diarylethenes 1–3 had good fatigue resistance in both hexane and PMMA films.
![[1860-5397-8-114-4]](/bjoc/content/figures/1860-5397-8-114-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fatigue resistance of diarylethenes 1–3 in hexane in air atmosphere at room temperature: (A) in hexane; (B) in PMMA films. Initial absorbance of the sample was fixed to 1.0.
Figure 4: Fatigue resistance of diarylethenes 1–3 in hexane in air atmosphere at room temperature: (A) in hex...
Fluorescence of diarylethenes 1–3
Fluorescence can be used not only in molecular-scale optoelectronics but also in digital photoswitching [22,53,54]. Like most of reported diarylethenes, diarylethenes 1–3 exhibited notable fluorescence in both hexane and PMMA films. Their fluorescence spectra were measured at room temperature with a Hitachi F-4500 spectrophotometer (Figure 5). In hexane, the emission peaks of 1o–3o were observed at 384, 387, and 389 nm, when excited at 307, 315, and 300 nm, whereas those in PMMA films were observed at 438, 441, and 427 nm, when excited at 334, 300, and 300 nm respectively. In comparison with those of 1o–3o in hexane, the fluorescence emission peaks of 1o–3o in PMMA films consistently exhibited a remarkable bathochromic shift. The emission intensity of the ortho-substituted derivative 3o was the strongest, while that of the para-substituted derivative 1o was the weakest in both hexane and PMMA films. Compared to the unsubstituted parent diarylethene, 1-(2-methylnaphth-1-yl)-2-[2-methyl-5-phenylthien-3-yl]perfluorocyclopentene (Φf = 0.011) [37], the fluorescence emission intensities of diarylethenes 1o and 2o were decreased, but that of 3o was evidently increased. When anthracene was used as the reference, the fluorescence quantum yields of 1o–3o were determined to be 0.012, 0.018, and 0.059, respectively, indicating that the formyl group on the terminal benzene ring could notably enhance the fluorescence quantum yield and remarkably influence the fluorescence emission intensity of diarylethenes with a naphthalene moiety.
![[1860-5397-8-114-5]](/bjoc/content/figures/1860-5397-8-114-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fluorescence emission spectra of diarylethenes 1–3 at room temperature: (A) in hexane solution (2.0 × 10−5 mol L−1); (B) in PMMA films (10%, w/w).
Figure 5: Fluorescence emission spectra of diarylethenes 1–3 at room temperature: (A) in hexane solution (2.0...
Diarylethenes 1–3 exhibited an evident fluorescence switching capability upon changing from the open-ring to the closed-ring isomers by photoirradiation in both hexane and PMMA films. When irradiated by UV light, the photocyclization reaction yielded the nonfluorescent closed-ring isomers 1c–3c, resulting in a decrease in emission intensity as compared to the open-ring isomers 1o–3o. The back irradiation by visible light of appropriate wavelength (λ > 500 nm) regenerated the open-ring isomers 1o–3o, and recovered their original emission intensity. As shown in Figure 6, upon irradiation with UV light, the emission intensity of 1 decreased, and was quenched to ca. 59% in hexane and 35% in a PMMA film, when it arrived at the photostationary state. The fluorescent modulation efficiency in the photostationary state was 41% in hexane and 65% in a PMMA film. Similarly, in the photostationary state, the fluorescence modulation efficiencies of diarylethenes 2 and 3 in hexane were 73 and 78%, and those in PMMA films were 38 and 42%, respectively. The residual fluorescence for 1–3 in the photostationary state may be attributed to an incomplete cyclization reaction and the existence of parallel conformations [55,56]. Among the three isomeric derivatives, the fluorescent modulation efficiency of diarylethene 2 was the largest and that of 1 was the smallest in PMMA films, suggesting that the diarylethene 2 is the best candidate for the fluorescence photoswitching material.
![[1860-5397-8-114-6]](/bjoc/content/figures/1860-5397-8-114-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Emission intensity changes of diarylethene 1 upon irradiation with UV light at room temperature: (A) in hexane (excited at 307 nm), (B) in a PMMA film (excited at 324 nm).
Figure 6: Emission intensity changes of diarylethene 1 upon irradiation with UV light at room temperature: (A...
Electrochemical properties of diarylethenes 1–3
The electrochemical behaviors of diarylethene derivatives have attracted much attention because of their potential applications in molecular-scale electronic switches [57-62]. The electrochemical properties of 1–3 were evaluated by cyclic voltammetry (CV) under the same experimental conditions reported previously [13]. The CV curves of diarylethenes 1–3 are shown in Figure 7. The onset potentials (Eonset) of oxidation and reduction for 1o were initiated at +1.79 and −0.89 V, and those of 1c at +1.76 and −0.91 V, respectively. According to the reported method [63,64], the ionization potential and electron affinities of 1o were calculated to be −6.59 and −4.09 eV, and those of 1c were −6.56 and −3.89 eV. Based on the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy level, the band gap Eg (Eg = LUMO–HOMO) of 1o and 1c can be determined to be +2.50 and +2.67 eV. Similarly, the oxidation potential of 2o and 3o is initiated at +1.76 and +1.83 V, and that of 2c and 3c is initiated at +1.74 and +1.79 V. The results indicate that the oxidation process for the open-ring isomers 1o–3o occurs at higher potentials than in the corresponding closed-ring isomers 1c–3c. This was because the longer conjugation length of the closed-ring isomers generally leads to a less positive potential [62,65]. The cyclization reaction allows the π-conjugation to extend across the perfluorocyclopentene ring causing a lower oxidation onset. As shown in Table 2, for the band gap of diarylethenes 1 and 2, the values of Eg of the open-ring isomers were lower than those of the closed-ring isomers, with the exception of 3. Among these compounds, the Eg of 1o was the smallest, which implies that the charge transfer of 1o was faster compared to that in others [66]. All these data suggest that the position of the formyl group at the terminal benzene ring has a remarkable effect on the electrochemical behaviors of these diarylethenes, but further work is required to quantify these effects.
![[1860-5397-8-114-7]](/bjoc/content/figures/1860-5397-8-114-7.png?scale=2.8&max-width=1024&background=FFFFFF)
Figure 7: Cyclic voltammetry of diarylethenes 1–3 in acetonitrile with a scanning rate of 50 mV/s.
Figure 7: Cyclic voltammetry of diarylethenes 1–3 in acetonitrile with a scanning rate of 50 mV/s.
Table 2: Electrochemical properties of diarylethenes 1–3.
| compound | oxidation | reduction | band gap | ||
|---|---|---|---|---|---|
| Eonset (V) | IP (eV) | Eonset (V) | EA (eV) | Eg | |
| 1o | +1.79 | −6.59 | −0.89 | −4.09 | 2.50 |
| 1c | +1.76 | −6.56 | −0.91 | −3.89 | 2.67 |
| 2o | +1.76 | −6.56 | −0.83 | −3.97 | 2.59 |
| 2c | +1.74 | −6.54 | −1.01 | −3.79 | 2.75 |
| 3o | +1.83 | −6.63 | −0.87 | −3.93 | 2.70 |
| 3c | +1.79 | −6.59 | −0.80 | −4.00 | 2.59 |
Conclusion
Three new asymmetric isomeric diarylethenes containing a formyl group at either the para, meta, or ortho position of the terminal benzene ring were synthesized for the investigation of the effect of substituent position on their optical and electrochemical properties. The results revealed that the formyl group and its position had significant effects on the properties of these isomeric diarylethenes. The electron-withdrawing formyl group endowed these diarylethenes with some new properties, which were different from those of diarylethenes with a methoxy group or halide at the terminal benzene.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectral data. | ||
| Format: PDF | Size: 331.2 KB | Download |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (20962008, 21162011), the Project of Jiangxi Academic and Technological Leader (2009DD00100), the Project of Jiangxi Youth Scientist, and the Project of the Science Funds of Jiangxi Education Office (GJJ10241, GJJ09646).
References
-
Feringa, B. L., Ed. Molecular Switches; Wiley-VCH: Weinheim, Germany, 2001. doi:10.1002/3527600329.fmatter_indsub
Return to citation in text: [1] [2] -
Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d
Return to citation in text: [1] [2] [3] [4] -
Irie, M. Diarylethenes with Heterocyclic Aryl Groups. In Organic Photochromic and Thermochromic Compounds: Physicochemical Studies, Biological Applications, and Thermochromism; Crano, J. C.; Guglielmetti, R. J., Eds.; Plenum Press: New York, 1999; Vol. 1, pp 207–222.
Return to citation in text: [1] -
Luo, Q.; Cheng, H.; Tian, H. Polym. Chem. 2011, 2, 2435–2443. doi:10.1039/c1py00167a
Return to citation in text: [1] -
Norsten, T. B.; Branda, N. R. J. Am. Chem. Soc. 2001, 123, 1784–1785. doi:10.1021/ja005639h
Return to citation in text: [1] -
Giordano, L.; Jovin, T. M.; Irie, M.; Jares-Erijman, E. A. J. Am. Chem. Soc. 2002, 124, 7481–7489. doi:10.1021/ja016969k
Return to citation in text: [1] -
Liddell, P. A.; Kodis, G.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 2002, 124, 7668–7669. doi:10.1021/ja026327c
Return to citation in text: [1] -
Yamaguchi, T.; Irie, M. J. Org. Chem. 2005, 70, 10323–10328. doi:10.1021/jo051372z
Return to citation in text: [1] -
Tsivgoulis, G. M.; Lehn, J.-M. Chem.–Eur. J. 1996, 2, 1399–1406. doi:10.1002/chem.19960021112
Return to citation in text: [1] -
Pu, S.; Yang, T.; Xu, J.; Chen, B. Tetrahedron Lett. 2006, 47, 6473–6477. doi:10.1016/j.tetlet.2006.06.073
Return to citation in text: [1] -
Fan, C.; Pu, S.; Liu, G.; Yang, T. J. Photochem. Photobiol., A: Chem. 2008, 194, 333–343. doi:10.1016/j.jphotochem.2007.08.032
Return to citation in text: [1] -
Pu, S.; Yan, L.; Wen, Z.; Liu, G.; Shen, L. J. Photochem. Photobiol., A: Chem. 2008, 196, 84–93. doi:10.1016/j.jphotochem.2007.11.016
Return to citation in text: [1] -
Pu, S.; Zheng, C.; Le, Z.; Liu, G.; Fan, C. Tetrahedron 2008, 64, 2576–2585. doi:10.1016/j.tet.2008.01.025
Return to citation in text: [1] [2] -
Pu, S.; Fan, C.; Miao, W.; Liu, G. Dyes Pigm. 2010, 84, 25–35. doi:10.1016/j.dyepig.2009.06.018
Return to citation in text: [1] [2] -
Goeldner, M.; Givens, R., Eds. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Gorostiza, P.; Isacoff, E. Y. Science 2008, 322, 395–399. doi:10.1126/science.1166022
Return to citation in text: [1] -
Zou, Q.; Li, X.; Zhang, J.; Zhou, J.; Sun, B.; Tian, H. Chem. Commun. 2012, 48, 2095–2097. doi:10.1039/c2cc16942h
Return to citation in text: [1] -
Finley, K. R.; Davidson, A. E.; Ekker, S. C. BioTechniques 2001, 31, 66–72.
http://www.biotechniques.com/multimedia/archive/00011/01311st02_11391a.pdf
Return to citation in text: [1] -
Zheng, Q.; Xu, G.; Prasad, P. N. Chem.–Eur. J. 2008, 14, 5812–5819. doi:10.1002/chem.200800309
Return to citation in text: [1] -
Mottram, L. F.; Maddox, E.; Schwab, M.; Beaufils, F.; Peterson, B. R. Org. Lett. 2007, 9, 3741–3744. doi:10.1021/ol7015093
Return to citation in text: [1] -
Miller, E. W.; Bian, S. X.; Chang, C. J. J. Am. Chem. Soc. 2007, 129, 3458–3459. doi:10.1021/ja0668973
Return to citation in text: [1] -
Tian, H.; Feng, Y. J. Mater. Chem. 2008, 18, 1617–1622. doi:10.1039/B713216F
Return to citation in text: [1] [2] -
Willner, I.; Rubin, S.; Wonner, J.; Effenberger, F.; Baeuerle, P. J. Am. Chem. Soc. 1992, 114, 3150–3151. doi:10.1021/ja00034a078
Return to citation in text: [1] -
Lion-Dagan, M.; Willner, I. J. Photochem. Photobiol., A: Chem. 1997, 108, 247–252. doi:10.1016/S1010-6030(97)00083-X
Return to citation in text: [1] -
Sakata, T.; Yan, Y.; Marriott, G. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4759–4764. doi:10.1073/pnas.0405265102
Return to citation in text: [1] -
Weston, D. G.; Kirkham, J.; Cullen, D. C. Biochim. Biophys. Acta 1999, 1428, 463–467. doi:10.1016/S0304-4165(99)00099-9
Return to citation in text: [1] -
Renner, C.; Moroder, L. ChemBioChem 2006, 7, 868–878. doi:10.1002/cbic.200500531
Return to citation in text: [1] -
Volgraf, M.; Gorostiza, P.; Numano, R.; Kramer, R. H.; Isacoff, E. Y.; Trauner, D. Nat. Chem. Biol. 2005, 2, 47–52. doi:10.1038/nchembio756
Return to citation in text: [1] -
Willner, I.; Rubin, S.; Riklin, A. J. Am. Chem. Soc. 1991, 113, 3321–3325. doi:10.1021/ja00009a016
Return to citation in text: [1] -
Woolley, G. A. Chem. Res. 2005, 38, 486–493. doi:10.1021/ar040091v
Return to citation in text: [1] -
Zhang, F.; Zarrine-Afsar, A.; Al-Abdul-Wahid, M. S.; Prosser, R. S.; Davidson, A. R.; Woolley, G. A. J. Am. Chem. Soc. 2009, 131, 2283–2289. doi:10.1021/ja807938v
Return to citation in text: [1] -
Zou, Y.; Yi, T.; Xiao, S.; Li, F.; Li, C.; Gao, X.; Wu, J.; Yu, M.; Huang, C. J. Am. Chem. Soc. 2008, 130, 15750–15751. doi:10.1021/ja8043163
Return to citation in text: [1] -
Piao, X.; Zou, Y.; Wu, J.; Li, C.; Yi, T. Org. Lett. 2009, 11, 3818–3821. doi:10.1021/ol9014267
Return to citation in text: [1] -
Singer, M.; Jäschke, A. J. Am. Chem. Soc. 2010, 132, 8372–8377. doi:10.1021/ja1024782
Return to citation in text: [1] -
Wu, Y.; Chen, S.; Yang, Y.; Zhang, Q.; Xie, Y.; Tian, H.; Zhu, W. Chem. Commun. 2012, 48, 528–530. doi:10.1039/c1cc15824d
Return to citation in text: [1] -
Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/B302356G
Return to citation in text: [1] -
Wang, R.; Pu, S.; Liu, G.; Liu, W.; Xia, H. Tetrahedron Lett. 2011, 52, 3306–3310. doi:10.1016/j.tetlet.2011.04.063
Return to citation in text: [1] [2] [3] -
Li, Z.-X.; Liao, L.-Y.; Sun, W.; Xu, C.-H.; Zhang, C.; Fang, C.-J.; Yan, C.-H. J. Phys. Chem. C 2008, 112, 5190–5196. doi:10.1021/jp711613y
Return to citation in text: [1] -
Yamamoto, S.; Matsuda, K.; Irie, M. Org. Lett. 2003, 5, 1769–1772. doi:10.1021/ol034440h
Return to citation in text: [1] [2] -
Yamamoto, S.; Matsuda, K.; Irie, M. Angew. Chem., Int. Ed. 2003, 42, 1636–1639. doi:10.1002/anie.200250417
Return to citation in text: [1] [2] -
Yamamoto, S.; Matsuda, K.; Irie, M. Chem.–Eur. J. 2003, 9, 4878–4886. doi:10.1002/chem.200304947
Return to citation in text: [1] [2] -
Kuroki, L.; Takami, S.; Shibata, K.; Irie, M. Chem. Commun. 2005, 6005–6007. doi:10.1039/B512873K
Return to citation in text: [1] -
Pu, S.; Li, M.; Liu, G.; Le, Z. Aust. J. Chem. 2009, 62, 464–474.
http://www.publish.csiro.au/paper/CH08289
Return to citation in text: [1] -
Wang, R. J.; Li, W. B.; Liu, W. J.; Liu, G. Adv. Mater. Res. 2012, 393–395, 381–384. doi:10.4028/www.scientific.net/AMR.393-395.381
Return to citation in text: [1] -
Liu, G.; Pu, S.; Wang, X. J. Photochem. Photobiol., A: Chem. 2010, 214, 230–240. doi:10.1016/j.jphotochem.2010.07.001
Return to citation in text: [1] -
Kawai, S.; Nakashima, T.; Atsumi, K.; Sakai, T.; Harigai, M.; Imamoto, Y.; Kamikubo, H.; Kataoka, M.; Kawai, T. Chem. Mater. 2007, 19, 3479–3483. doi:10.1021/cm0630340
Return to citation in text: [1] -
Pu, S.; Li, H.; Liu, G.; Liu, W.; Cui, S.; Fan, C. Tetrahedron 2011, 67, 1438–1447. doi:10.1016/j.tet.2010.12.041
Return to citation in text: [1] -
Liu, G.; Pu, S.; Wang, X. Tetrahedron 2010, 66, 8862–8871. doi:10.1016/j.tet.2010.09.066
Return to citation in text: [1] -
Pu, S.; Liu, W.; Liu, G. Dyes Pigm. 2010, 87, 1–9. doi:10.1016/j.dyepig.2010.01.015
Return to citation in text: [1] [2] -
Pu, S.; Liu, W.; Miao, W. J. Phys. Org. Chem. 2009, 22, 954–963. doi:10.1002/poc.1545
Return to citation in text: [1] [2] -
Liu, G.; Pu, S.; Wang, X.; Liu, W.; Fan, C. Dyes Pigm. 2011, 90, 89–99. doi:10.1016/j.dyepig.2010.12.007
Return to citation in text: [1] -
Tian, H.; Wang, S. Chem. Commun. 2007, 781–792. doi:10.1039/B610004J
Return to citation in text: [1] -
Fukaminato, T.; Sasaki, T.; Kawai, T.; Tamai, N.; Irie, M. J. Am. Chem. Soc. 2004, 126, 14843–14849. doi:10.1021/ja047169n
Return to citation in text: [1] -
Zhang, J.; Tan, W.; Meng, X.; Tian, H. J. Mater. Chem. 2009, 19, 5726–5729. doi:10.1039/B908707A
Return to citation in text: [1] -
Fan, C.; Pu, S.; Liu, G.; Yang, T. J. Photochem. Photobiol., A: Chem. 2008, 197, 415–425. doi:10.1016/j.jphotochem.2008.02.004
Return to citation in text: [1] -
Pu, S.; Fan, C.; Miao, W.; Liu, G. Tetrahedron 2008, 64, 9464–9470. doi:10.1016/j.tet.2008.07.073
Return to citation in text: [1] -
Bonifazi, D.; Scholl, M.; Song, F.; Echegoyen, L.; Accorsi, G.; Armaroli, N.; Diederich, F. Angew. Chem., Int. Ed. 2003, 42, 4966–4970. doi:10.1002/anie.200352265
Return to citation in text: [1] -
Browne, W. R.; de Jong, J. J. D.; Kudernac, T.; Walko, M.; Lucas, L. N.; Uchida, K.; van Esch, J. H.; Feringa, B. L. Chem.–Eur. J. 2005, 11, 6414–6429. doi:10.1002/chem.200500162
Return to citation in text: [1] -
Browne, W. R.; de Jong, J. J. D.; Kudernac, T.; Walko, M.; Lucas, L. N.; Uchida, K.; van Esch, J. H.; Feringa, B. L. Chem.–Eur. J. 2005, 11, 6430–6441. doi:10.1002/chem.200500163
Return to citation in text: [1] -
Moriyama, Y.; Matsuda, K.; Tanifuji, N.; Irie, S.; Irie, M. Org. Lett. 2005, 7, 3315–3318. doi:10.1021/ol051149o
Return to citation in text: [1] -
Zheng, C.; Pu, S.; Xu, J.; Luo, M.; Huang, D.; Shen, L. Tetrahedron 2007, 63, 5437–5449. doi:10.1016/j.tet.2007.04.049
Return to citation in text: [1] -
Pu, S.; Yang, T.; Xu, J.; Shen, L.; Li, G.; Xiao, Q.; Chen, B. Tetrahedron 2005, 61, 6623–6629. doi:10.1016/j.tet.2005.04.044
Return to citation in text: [1] [2] -
Tsai, F.-C.; Chang, C.-C.; Liu, C.-L.; Chen, W.-C.; Jenekhe, S. A. Macromolecules 2005, 38, 1958–1966. doi:10.1021/ma048112o
Return to citation in text: [1] -
Zhan, X.; Liu, Y.; Wu, X.; Wang, S.; Zhu, D. Macromolecules 2002, 35, 2529–2537. doi:10.1021/ma011593g
Return to citation in text: [1] -
Perrier, A.; Maurel, F.; Aubard, J. J. Photochem. Photobiol., A: Chem. 2007, 189, 167–176. doi:10.1016/j.jphotochem.2007.01.030
Return to citation in text: [1] -
Kim, E.; Kim, M.; Kim, K. Tetrahedron 2006, 62, 6814–6821. doi:10.1016/j.tet.2006.04.089
Return to citation in text: [1]
| 63. | Tsai, F.-C.; Chang, C.-C.; Liu, C.-L.; Chen, W.-C.; Jenekhe, S. A. Macromolecules 2005, 38, 1958–1966. doi:10.1021/ma048112o |
| 64. | Zhan, X.; Liu, Y.; Wu, X.; Wang, S.; Zhu, D. Macromolecules 2002, 35, 2529–2537. doi:10.1021/ma011593g |
| 62. | Pu, S.; Yang, T.; Xu, J.; Shen, L.; Li, G.; Xiao, Q.; Chen, B. Tetrahedron 2005, 61, 6623–6629. doi:10.1016/j.tet.2005.04.044 |
| 65. | Perrier, A.; Maurel, F.; Aubard, J. J. Photochem. Photobiol., A: Chem. 2007, 189, 167–176. doi:10.1016/j.jphotochem.2007.01.030 |
| 66. | Kim, E.; Kim, M.; Kim, K. Tetrahedron 2006, 62, 6814–6821. doi:10.1016/j.tet.2006.04.089 |
| 1. | Feringa, B. L., Ed. Molecular Switches; Wiley-VCH: Weinheim, Germany, 2001. doi:10.1002/3527600329.fmatter_indsub |
| 2. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 23. | Willner, I.; Rubin, S.; Wonner, J.; Effenberger, F.; Baeuerle, P. J. Am. Chem. Soc. 1992, 114, 3150–3151. doi:10.1021/ja00034a078 |
| 24. | Lion-Dagan, M.; Willner, I. J. Photochem. Photobiol., A: Chem. 1997, 108, 247–252. doi:10.1016/S1010-6030(97)00083-X |
| 25. | Sakata, T.; Yan, Y.; Marriott, G. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4759–4764. doi:10.1073/pnas.0405265102 |
| 26. | Weston, D. G.; Kirkham, J.; Cullen, D. C. Biochim. Biophys. Acta 1999, 1428, 463–467. doi:10.1016/S0304-4165(99)00099-9 |
| 27. | Renner, C.; Moroder, L. ChemBioChem 2006, 7, 868–878. doi:10.1002/cbic.200500531 |
| 28. | Volgraf, M.; Gorostiza, P.; Numano, R.; Kramer, R. H.; Isacoff, E. Y.; Trauner, D. Nat. Chem. Biol. 2005, 2, 47–52. doi:10.1038/nchembio756 |
| 29. | Willner, I.; Rubin, S.; Riklin, A. J. Am. Chem. Soc. 1991, 113, 3321–3325. doi:10.1021/ja00009a016 |
| 30. | Woolley, G. A. Chem. Res. 2005, 38, 486–493. doi:10.1021/ar040091v |
| 31. | Zhang, F.; Zarrine-Afsar, A.; Al-Abdul-Wahid, M. S.; Prosser, R. S.; Davidson, A. R.; Woolley, G. A. J. Am. Chem. Soc. 2009, 131, 2283–2289. doi:10.1021/ja807938v |
| 44. | Wang, R. J.; Li, W. B.; Liu, W. J.; Liu, G. Adv. Mater. Res. 2012, 393–395, 381–384. doi:10.4028/www.scientific.net/AMR.393-395.381 |
| 15. | Goeldner, M.; Givens, R., Eds. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules; Wiley-VCH: Weinheim, Germany, 2005. |
| 16. | Gorostiza, P.; Isacoff, E. Y. Science 2008, 322, 395–399. doi:10.1126/science.1166022 |
| 17. | Zou, Q.; Li, X.; Zhang, J.; Zhou, J.; Sun, B.; Tian, H. Chem. Commun. 2012, 48, 2095–2097. doi:10.1039/c2cc16942h |
| 18. |
Finley, K. R.; Davidson, A. E.; Ekker, S. C. BioTechniques 2001, 31, 66–72.
http://www.biotechniques.com/multimedia/archive/00011/01311st02_11391a.pdf |
| 19. | Zheng, Q.; Xu, G.; Prasad, P. N. Chem.–Eur. J. 2008, 14, 5812–5819. doi:10.1002/chem.200800309 |
| 20. | Mottram, L. F.; Maddox, E.; Schwab, M.; Beaufils, F.; Peterson, B. R. Org. Lett. 2007, 9, 3741–3744. doi:10.1021/ol7015093 |
| 21. | Miller, E. W.; Bian, S. X.; Chang, C. J. J. Am. Chem. Soc. 2007, 129, 3458–3459. doi:10.1021/ja0668973 |
| 22. | Tian, H.; Feng, Y. J. Mater. Chem. 2008, 18, 1617–1622. doi:10.1039/B713216F |
| 45. | Liu, G.; Pu, S.; Wang, X. J. Photochem. Photobiol., A: Chem. 2010, 214, 230–240. doi:10.1016/j.jphotochem.2010.07.001 |
| 4. | Luo, Q.; Cheng, H.; Tian, H. Polym. Chem. 2011, 2, 2435–2443. doi:10.1039/c1py00167a |
| 5. | Norsten, T. B.; Branda, N. R. J. Am. Chem. Soc. 2001, 123, 1784–1785. doi:10.1021/ja005639h |
| 6. | Giordano, L.; Jovin, T. M.; Irie, M.; Jares-Erijman, E. A. J. Am. Chem. Soc. 2002, 124, 7481–7489. doi:10.1021/ja016969k |
| 7. | Liddell, P. A.; Kodis, G.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 2002, 124, 7668–7669. doi:10.1021/ja026327c |
| 8. | Yamaguchi, T.; Irie, M. J. Org. Chem. 2005, 70, 10323–10328. doi:10.1021/jo051372z |
| 9. | Tsivgoulis, G. M.; Lehn, J.-M. Chem.–Eur. J. 1996, 2, 1399–1406. doi:10.1002/chem.19960021112 |
| 10. | Pu, S.; Yang, T.; Xu, J.; Chen, B. Tetrahedron Lett. 2006, 47, 6473–6477. doi:10.1016/j.tetlet.2006.06.073 |
| 11. | Fan, C.; Pu, S.; Liu, G.; Yang, T. J. Photochem. Photobiol., A: Chem. 2008, 194, 333–343. doi:10.1016/j.jphotochem.2007.08.032 |
| 12. | Pu, S.; Yan, L.; Wen, Z.; Liu, G.; Shen, L. J. Photochem. Photobiol., A: Chem. 2008, 196, 84–93. doi:10.1016/j.jphotochem.2007.11.016 |
| 13. | Pu, S.; Zheng, C.; Le, Z.; Liu, G.; Fan, C. Tetrahedron 2008, 64, 2576–2585. doi:10.1016/j.tet.2008.01.025 |
| 14. | Pu, S.; Fan, C.; Miao, W.; Liu, G. Dyes Pigm. 2010, 84, 25–35. doi:10.1016/j.dyepig.2009.06.018 |
| 39. | Yamamoto, S.; Matsuda, K.; Irie, M. Org. Lett. 2003, 5, 1769–1772. doi:10.1021/ol034440h |
| 40. | Yamamoto, S.; Matsuda, K.; Irie, M. Angew. Chem., Int. Ed. 2003, 42, 1636–1639. doi:10.1002/anie.200250417 |
| 41. | Yamamoto, S.; Matsuda, K.; Irie, M. Chem.–Eur. J. 2003, 9, 4878–4886. doi:10.1002/chem.200304947 |
| 43. |
Pu, S.; Li, M.; Liu, G.; Le, Z. Aust. J. Chem. 2009, 62, 464–474.
http://www.publish.csiro.au/paper/CH08289 |
| 1. | Feringa, B. L., Ed. Molecular Switches; Wiley-VCH: Weinheim, Germany, 2001. doi:10.1002/3527600329.fmatter_indsub |
| 2. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 3. | Irie, M. Diarylethenes with Heterocyclic Aryl Groups. In Organic Photochromic and Thermochromic Compounds: Physicochemical Studies, Biological Applications, and Thermochromism; Crano, J. C.; Guglielmetti, R. J., Eds.; Plenum Press: New York, 1999; Vol. 1, pp 207–222. |
| 37. | Wang, R.; Pu, S.; Liu, G.; Liu, W.; Xia, H. Tetrahedron Lett. 2011, 52, 3306–3310. doi:10.1016/j.tetlet.2011.04.063 |
| 35. | Wu, Y.; Chen, S.; Yang, Y.; Zhang, Q.; Xie, Y.; Tian, H.; Zhu, W. Chem. Commun. 2012, 48, 528–530. doi:10.1039/c1cc15824d |
| 37. | Wang, R.; Pu, S.; Liu, G.; Liu, W.; Xia, H. Tetrahedron Lett. 2011, 52, 3306–3310. doi:10.1016/j.tetlet.2011.04.063 |
| 34. | Singer, M.; Jäschke, A. J. Am. Chem. Soc. 2010, 132, 8372–8377. doi:10.1021/ja1024782 |
| 38. | Li, Z.-X.; Liao, L.-Y.; Sun, W.; Xu, C.-H.; Zhang, C.; Fang, C.-J.; Yan, C.-H. J. Phys. Chem. C 2008, 112, 5190–5196. doi:10.1021/jp711613y |
| 39. | Yamamoto, S.; Matsuda, K.; Irie, M. Org. Lett. 2003, 5, 1769–1772. doi:10.1021/ol034440h |
| 40. | Yamamoto, S.; Matsuda, K.; Irie, M. Angew. Chem., Int. Ed. 2003, 42, 1636–1639. doi:10.1002/anie.200250417 |
| 41. | Yamamoto, S.; Matsuda, K.; Irie, M. Chem.–Eur. J. 2003, 9, 4878–4886. doi:10.1002/chem.200304947 |
| 42. | Kuroki, L.; Takami, S.; Shibata, K.; Irie, M. Chem. Commun. 2005, 6005–6007. doi:10.1039/B512873K |
| 33. | Piao, X.; Zou, Y.; Wu, J.; Li, C.; Yi, T. Org. Lett. 2009, 11, 3818–3821. doi:10.1021/ol9014267 |
| 32. | Zou, Y.; Yi, T.; Xiao, S.; Li, F.; Li, C.; Gao, X.; Wu, J.; Yu, M.; Huang, C. J. Am. Chem. Soc. 2008, 130, 15750–15751. doi:10.1021/ja8043163 |
| 2. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 36. | Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/B302356G |
| 49. | Pu, S.; Liu, W.; Liu, G. Dyes Pigm. 2010, 87, 1–9. doi:10.1016/j.dyepig.2010.01.015 |
| 46. | Kawai, S.; Nakashima, T.; Atsumi, K.; Sakai, T.; Harigai, M.; Imamoto, Y.; Kamikubo, H.; Kataoka, M.; Kawai, T. Chem. Mater. 2007, 19, 3479–3483. doi:10.1021/cm0630340 |
| 47. | Pu, S.; Li, H.; Liu, G.; Liu, W.; Cui, S.; Fan, C. Tetrahedron 2011, 67, 1438–1447. doi:10.1016/j.tet.2010.12.041 |
| 48. | Liu, G.; Pu, S.; Wang, X. Tetrahedron 2010, 66, 8862–8871. doi:10.1016/j.tet.2010.09.066 |
| 57. | Bonifazi, D.; Scholl, M.; Song, F.; Echegoyen, L.; Accorsi, G.; Armaroli, N.; Diederich, F. Angew. Chem., Int. Ed. 2003, 42, 4966–4970. doi:10.1002/anie.200352265 |
| 58. | Browne, W. R.; de Jong, J. J. D.; Kudernac, T.; Walko, M.; Lucas, L. N.; Uchida, K.; van Esch, J. H.; Feringa, B. L. Chem.–Eur. J. 2005, 11, 6414–6429. doi:10.1002/chem.200500162 |
| 59. | Browne, W. R.; de Jong, J. J. D.; Kudernac, T.; Walko, M.; Lucas, L. N.; Uchida, K.; van Esch, J. H.; Feringa, B. L. Chem.–Eur. J. 2005, 11, 6430–6441. doi:10.1002/chem.200500163 |
| 60. | Moriyama, Y.; Matsuda, K.; Tanifuji, N.; Irie, S.; Irie, M. Org. Lett. 2005, 7, 3315–3318. doi:10.1021/ol051149o |
| 61. | Zheng, C.; Pu, S.; Xu, J.; Luo, M.; Huang, D.; Shen, L. Tetrahedron 2007, 63, 5437–5449. doi:10.1016/j.tet.2007.04.049 |
| 62. | Pu, S.; Yang, T.; Xu, J.; Shen, L.; Li, G.; Xiao, Q.; Chen, B. Tetrahedron 2005, 61, 6623–6629. doi:10.1016/j.tet.2005.04.044 |
| 13. | Pu, S.; Zheng, C.; Le, Z.; Liu, G.; Fan, C. Tetrahedron 2008, 64, 2576–2585. doi:10.1016/j.tet.2008.01.025 |
| 37. | Wang, R.; Pu, S.; Liu, G.; Liu, W.; Xia, H. Tetrahedron Lett. 2011, 52, 3306–3310. doi:10.1016/j.tetlet.2011.04.063 |
| 55. | Fan, C.; Pu, S.; Liu, G.; Yang, T. J. Photochem. Photobiol., A: Chem. 2008, 197, 415–425. doi:10.1016/j.jphotochem.2008.02.004 |
| 56. | Pu, S.; Fan, C.; Miao, W.; Liu, G. Tetrahedron 2008, 64, 9464–9470. doi:10.1016/j.tet.2008.07.073 |
| 2. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 52. | Tian, H.; Wang, S. Chem. Commun. 2007, 781–792. doi:10.1039/B610004J |
| 22. | Tian, H.; Feng, Y. J. Mater. Chem. 2008, 18, 1617–1622. doi:10.1039/B713216F |
| 53. | Fukaminato, T.; Sasaki, T.; Kawai, T.; Tamai, N.; Irie, M. J. Am. Chem. Soc. 2004, 126, 14843–14849. doi:10.1021/ja047169n |
| 54. | Zhang, J.; Tan, W.; Meng, X.; Tian, H. J. Mater. Chem. 2009, 19, 5726–5729. doi:10.1039/B908707A |
| 14. | Pu, S.; Fan, C.; Miao, W.; Liu, G. Dyes Pigm. 2010, 84, 25–35. doi:10.1016/j.dyepig.2009.06.018 |
| 50. | Pu, S.; Liu, W.; Miao, W. J. Phys. Org. Chem. 2009, 22, 954–963. doi:10.1002/poc.1545 |
| 51. | Liu, G.; Pu, S.; Wang, X.; Liu, W.; Fan, C. Dyes Pigm. 2011, 90, 89–99. doi:10.1016/j.dyepig.2010.12.007 |
| 49. | Pu, S.; Liu, W.; Liu, G. Dyes Pigm. 2010, 87, 1–9. doi:10.1016/j.dyepig.2010.01.015 |
| 50. | Pu, S.; Liu, W.; Miao, W. J. Phys. Org. Chem. 2009, 22, 954–963. doi:10.1002/poc.1545 |
© 2012 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)