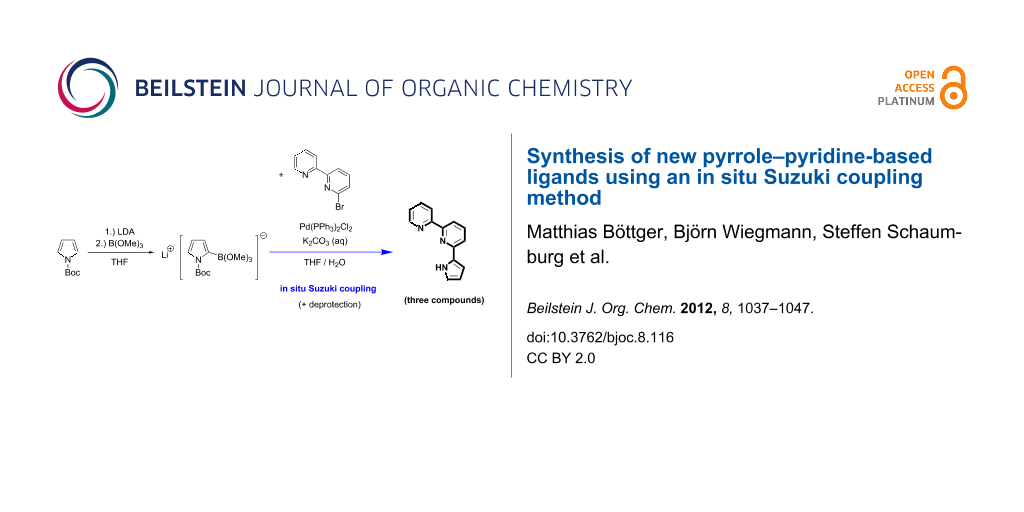
Abstract
The compounds 6-(pyrrol-2-yl)-2,2‘-bipyridine, 2-(pyrrol-2-yl)-1,10-phenanthroline and 2-(2-(N-methylbenz[d,e]imidazole)-6-(pyrrol-2-yl)-pyridine were synthesized by using an in situ generated boronic acid for the Suzuki coupling. Crystals of the products could be grown and exhibited interesting structures by X-ray analysis, one of them showing a chain-like network with the adjacent molecules linked to each other via intermolecular N–H…N hydrogen bonds.

Graphical Abstract
Introduction
Tridentate ligands have recently received attention in the area of rare-earth-metal complex chemistry [1-5]. Many rare-earth-metal cations, such as europium(III), have the tendency to be nine-coordinating species. Besides their triple positive charge, this coordination sphere must be saturated to achieve high quantum yields when the target is to optimize its luminescent properties [6]. In the “classic” complexes, i.e., the β-diketonates [6,7], this is often achieved by combining three “saturating ligands” that consist of negatively charged β-diketonates with a “neutral ligand”, which is often bidentate or tridentate (Scheme 1, left). The coordination sphere and the positive charge of the rare-earth metal cation are thus saturated and neutralized, respectively. In a recent development, the Bünzli group has succeeded in synthesizing homoleptic complexes, in which only one type of ligand binds to the central europium(III) via two neutral and one negatively charged atom (Scheme 1, middle) [2-4]. As three ligands bind to the rare-earth cation, the resulting complex is neutral, and its coordination number is nine and thereby saturated.
![[1860-5397-8-116-i1]](/bjoc/content/inline/1860-5397-8-116-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: β-diketonate complexes (left), homoleptic complexes (middle) and planned homoleptic complexes of europium(III) (right).
Scheme 1: β-diketonate complexes (left), homoleptic complexes (middle) and planned homoleptic complexes of eu...
Our interest in this type of complex is driven by its potential analytical application for luminescence degradation measurements on photocatalytic surfaces [8]. Currently, β-diketonate complexes are in use for this analysis, but homoleptic complexes may be advantageous in our opinion, because they only consist of one type of ligand, which is then subjected to photocatalytically produced radicals. Therefore, we tried to broaden the scope by synthesizing a new class of ligands for homoleptic complexes, which should bind to europium(III) via a pyrrolate anion (Scheme 1, right). The decision to choose a negatively charged heterocycle as a binding unit was based on the idea to enlarge the π-system of the ligand, thereby making it possible to absorb longer wavelengths of light (λ > 350 nm). The target structures are shown in Scheme 2.
![[1860-5397-8-116-i2]](/bjoc/content/inline/1860-5397-8-116-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Pyrrole–pyridine-based structures synthesized in this study.
Scheme 2: Pyrrole–pyridine-based structures synthesized in this study.
Structures 1 and 2 comprise substructures of common neutral ligands used in europium complex chemistry [6,9]: 2,2’-bipyridine and 1,10-phenanthroline. Compound 3 comprises a benzimidazole heterocycle, which was also used by the Bünzli group [1-4]. The synthesis of the resulting complexes was to date unsuccessful. We report here on the synthesis of the new structures 1–3.
Results and Discussion
Our first retrosynthetic approach included a Suzuki coupling of the alpha-substituted boronic acid of Boc-protected pyrrole 7 with the heteroaryl bromides 8–10, as shown in Scheme 3.
![[1860-5397-8-116-i3]](/bjoc/content/inline/1860-5397-8-116-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Retrosynthetic approach for structures 1–3.
Scheme 3: Retrosynthetic approach for structures 1–3.
Compound 7 is described in literature [10], but it could not be purified by column chromatography and therefore was not isolated as a pure product. In addition, reports on the stability of this boronic acid show that it is not suitable for long-term storage [10]. We therefore applied a modification of the Suzuki coupling that was also used to prepare [2.2]paracyclophane-derivatives [11]. This comprised the in situ reaction of the freshly prepared boronic acid/ester with the heteroaryl bromides 8–10. These starting compounds could be prepared by using literature procedures, as shown in Scheme 4.
![[1860-5397-8-116-i4]](/bjoc/content/inline/1860-5397-8-116-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the heteroaryl bromides used in the coupling reaction.
Scheme 4: Synthesis of the heteroaryl bromides used in the coupling reaction.
Substance 8 was synthesized by following the standard literature procedure [12]. Compound 9 was synthesized by principally the same reaction path [12,13]. 2-Bromopyridine-6-benzimidazole 10 was published in a patent [14], and its preparation was adapted from [4]. The following in situ Suzuki coupling uses Boc-protected pyrrole [15], which was subjected to freshly prepared LDA at −78 °C and quenched with trimethylborate. This gave rise to the intermediate 21/22 (Scheme 5).
![[1860-5397-8-116-i5]](/bjoc/content/inline/1860-5397-8-116-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Generation of the borate intermediate 21/22.
Scheme 5: Generation of the borate intermediate 21/22.
Afterwards the heteroaryl bromides 8–10 were added along with the catalyst and base in aqueous media and the reaction mixture was heated under reflux for several hours. The reaction times were not minimized. In principal the ester can react with the boronic acid, starting with the addition of the aqueous base (K2CO3). Yet, structure 21 is already activated for the coupling step, since a negative charge is located on the boron atom. In Scheme 6 the coupling reactions are shown; detailed information about the reaction conditions is given in Table 1.
![[1860-5397-8-116-i6]](/bjoc/content/inline/1860-5397-8-116-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: In situ Suzuki coupling reactions of the heteroaryl bromides 8–10.
Scheme 6: In situ Suzuki coupling reactions of the heteroaryl bromides 8–10.
Table 1: Reaction conditions used for coupling reactions.
| aryl bromide | 19 | LDA | B(OMe)3 | stirring timea | Pd(PPh3)Cl2 | K2CO3 (1 M) | product | yield | |
|---|---|---|---|---|---|---|---|---|---|
| [equiv] | [equiv] | [equiv] | [equiv] | [mol %] | [equiv] | ||||
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-116-i7.svg?max-width=637&scale=1.0)
8 |
1.2 | 1.0 | 1.1 | 1.5 | 15 h | 8 | 1.65 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-116-i8.svg?max-width=637&scale=1.0)
4 |
84% |
| 1.2 | 1.0 | 1.1 | 1.5 | 15 h | 8 | 1.65 | 87% | ||
| 1.0 | 1.25 | 1.4 | 1.9 | 1.5 h | 8 | 2.0 | 89% | ||
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-116-i9.svg?max-width=637&scale=1.0)
9 |
1.1 | 1.0 | 1.1 | 1.5 | 1.5 h | 8 | 1.65 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-116-i10.svg?max-width=637&scale=1.0)
2 |
41% |
| 1.1 | 1.0 | 1.1 | 1.5 | 16 h | 8 | 1.65 | 39% | ||
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-116-i11.svg?max-width=637&scale=1.0)
10 |
1.1 | 1.0 | 1.1 | 1.5 | 0.5 h | 5 | 1.65 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-116-i12.svg?max-width=637&scale=1.0)
6 |
52% |
| 1.0 | 1.3 | 1.25 | 1.3 | 0.5 h | 5 | 1.6 | 85% | ||
| 1.0 | 1.5 | 1.55 | 1.6 | 1.2 h | 5 | 2.0 | 87% | ||
aStirring time: After addition of B(OMe)3 at 0 °C to RT.
Reaction 1 of aryl bromide 8 to product 4 was repeated under the same conditions and with the same amounts of starting material and catalyst. The workup procedure was changed, which affected the yield only to a very small extent. In the original publication [11] the aryl bromide coupling partner was used in excess. As 19 is by far easier to prepare in large quantities, we altered the protocol and used it in excess, which produced even a slightly better yield than the other way round (Table 1). Finally, deprotection of 4 with hydrochloric acid after the coupling gave 1 in 93% yield. Compound 2 was directly isolated as Boc-deprotected product, but in a much lower yield. We believe the lower yield is caused by the workup procedure, because TLC indicated nearly full consumption of 19. During column purification with aluminium oxide or silica (both were tried) we noticed smearing of the blue fluorescent product under UV irradiation. Even deactivated aluminium oxide provides enough acidity to split the Boc-moiety, which probably causes the generated amine to partly remain on the column. In the case of 6, using an excess of 19 lead to better yields, but it has to be mentioned that the workup procedure was also adjusted, thus it cannot be directly compared. Further increasing the equivalents of 19 and the base did not significantly enhance the resulting yield (Table 1). The total reaction time was not systematically minimized. After 20 h under reflux 6 was isolated in 87% yield; a reaction time of 70 h delivered 85% of 6. Compound 3 was synthesized in 70% yield by deprotection of 6. In this case another method using sodium methoxide was applied, because reaction with hydrochloric acid did not take place and the unprotected starting material was recovered. We noticed no definite influence on the product yield when using different scales (2 mmol to 12 mmol).
X-Ray analysis
The molecular structure of compound 1 is shown in Figure 1. The angles subtended to the central ring are 30° from the pyridyl and 8° from the pyrrolyl substituent. The N…N configurations are trans for the bipyridyl substructure (N–C–C–N torsion angle −155.8(1)°) but cis for the pyrrolylpyridine substructure (torsion angle 3.8(2)°). The former is well-known as a structural preference of 2,2'-bipyridyl systems. A search of the Cambridge Structural Database [16] for 2,2'-pyrrolylpyridines revealed 20 hits, all with cis geometry, discounting rigid fused-ring systems and one sterically hindered di-tert-butyl system; the corresponding absolute torsion angles ranged from 0 to 25°, mean value 7.5°. The molecular packing of 1 (Figure 2) is determined by a classical hydrogen bond N17–H17…N1 involving the peripheral rings, which connects the molecules via the a-glide plane to form chains parallel to the a-axis.
![[1860-5397-8-116-1]](/bjoc/content/figures/1860-5397-8-116-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structure of compound 1 in the crystal. Ellipsoids correspond to 50% probability levels.
Figure 1: The structure of compound 1 in the crystal. Ellipsoids correspond to 50% probability levels.
![[1860-5397-8-116-2]](/bjoc/content/figures/1860-5397-8-116-2.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 2: Packing diagram of compound 1, viewed parallel to the y-axis in the range y ≈ 1/4. Hydrogen bonds are indicated by dashed lines. Hydrogen atoms not involved in the hydrogen bonds are omitted for clarity. Hydrogen bond details (Å,°) for N17–H17…N1: H…N 0.88(2), N…N 2.861(2), N–H…N 158(2), operator 1/2 + x, 1 1/2 − y, z.
Figure 2: Packing diagram of compound 1, viewed parallel to the y-axis in the range y ≈ 1/4. Hydrogen bonds a...
The structure of the methanol solvate of compound 2 is shown in Figure 3. The 1,10-phenanthroline ring system is planar (mean deviation 0.01 Å), and the pyrrole ring subtends an interplanar angle of 9° to it. The methanol molecule fits neatly into the “bay” region of the parent molecule, forming classical hydrogen bonds N19–H19…O99 and O99–H99…N1. The molecules associate into pairs by ring stacking across inversion centres (Figure 4). The interplanar distance is ca. 3.35 Å.
![[1860-5397-8-116-3]](/bjoc/content/figures/1860-5397-8-116-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The structure of compound 2·CH3OH in the crystal. Ellipsoids correspond to 50% probability levels. Hydrogen bond details (Å,°) for N19–H19…O99: H…O 1.96(2), N…O 2.839(1), N–H…O 174(1); for O99–H99…N1: H…N 1.93(2), O…N 2.841(1), N–H…O 173(2).
Figure 3: The structure of compound 2·CH3OH in the crystal. Ellipsoids correspond to 50% probability levels. ...
![[1860-5397-8-116-4]](/bjoc/content/figures/1860-5397-8-116-4.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 4: Packing diagram of compound 2·CH3OH showing the formation of inversion-symmetric "stacked" dimers. Hydrogen bonds are shown as thin dashed lines. Hydrogen atoms are omitted for clarity.
Figure 4: Packing diagram of compound 2·CH3OH showing the formation of inversion-symmetric "stacked" dimers. ...
The structure of the ethanol solvate of compound 3 is shown in Figure 5. The central ring is almost coplanar with the pyrrole ring (interplanar angle 9°), but subtends an angle of 38° with the benzimidazole system. Ethanol forms one classical hydrogen bond within the asymmetric unit, acting as a donor, but it also acts as a hydrogen bond acceptor via the a-glide plane. The overall effect is to form helical chains of alternating residues of 3 and ethanol, parallel to the a-axis (Figure 6).
![[1860-5397-8-116-5]](/bjoc/content/figures/1860-5397-8-116-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The structure of compound 3·C2H5OH in the crystal. Ellipsoids correspond to 50% probability levels. Hydrogen bond details (Å,°) for O99–H99…N17: H…N 1.91(2), O…N 2.813(1), O–H…N 174(2).
Figure 5: The structure of compound 3·C2H5OH in the crystal. Ellipsoids correspond to 50% probability levels....
![[1860-5397-8-116-6]](/bjoc/content/figures/1860-5397-8-116-6.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 6: Packing diagram of compound 3·C2H5OH. Hydrogen bonds are shown as thick dashed lines. Hydrogen atoms not involved in the hydrogen bonds are omitted for clarity. Hydrogen bond details (Å,°) for N1–H01…O99: H…O 2.01(2), N…O 2.848(1), N–H…O 154(1), operator −1/2 + x, y, 1/2 − z.
Figure 6: Packing diagram of compound 3·C2H5OH. Hydrogen bonds are shown as thick dashed lines. Hydrogen atom...
The crystallographic data of compounds 1 to 3 are summarized in Table 2.
Table 2: Crystallographic data for compounds 1, 2·CH3OH, 3·C2H5OH.
| compound | 1 | 2·CH3OH | 3·C2H5OH |
|---|---|---|---|
| formula | C14H11N3 | C17H15N3O | C19H20N4O |
| Mr | 221.26 | 277.32 | 320.39 |
| habit | colourless tablet | amber tablet | colourless block |
| crystallite size (mm) | 0.4 × 0.25 × 0.2 | 0.25 × 0.2 × 0.15 | 0.4 × 0.3 × 0.15 |
| crystal system | orthorhombic | monoclinic | orthorhombic |
| space group | Pna21 | P21/c | Pbca |
| cell constants: | |||
| a [Å] | 9.8842(4) | 9.7060(3) | 9.09323(15) |
| b [Å] | 16.1917(6) | 10.3442(3) | 17.5182(3) |
| c [Å] | 7.1501(3) | 13.4884(4) | 21.2257(4) |
| α [°] | 90 | 90 | 90 |
| β [°] | 90 | 91.043(4) | 90 |
| γ [°] | 90 | 90 | 90 |
| V (Å3) | 1144.32 | 1354.04 | 3381.19 |
| Z | 4 | 4 | 8 |
| Dx (g·cm−3) | 1.284 | 1.360 | 1.259 |
| μ (mm−1) | 0.08 | 0.09 | 0.08 |
| F(000) | 464 | 584 | 1360 |
| T (°C) | −173 | −173 | −173 |
| wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| 2θmax | 60 | 60 | 60 |
| Refl. measured | 34078 | 50772 | 125784 |
| Refl. indep. | 1791 | 3936 | 4949 |
| Rint | 0.029 | 0.028 | 0.034 |
| parameters | 158 | 100 | 240 |
| wR(F2, all refl.) | 0.084 | 0.111 | 0.104 |
| R(F, >4σ(F)) | 0.031 | 0.039 | 0.0406 |
| S | 1.01 | 1.03 | 1.04 |
| max. Δ/ρ (e·Å−3) | 0.25 | 0.36 | 0.36 |
Conclusion
The new target structures 1–3 were successfully synthesized in good to acceptable yields by applying an in situ variation of the Suzuki coupling as the main reaction step. The crystal structures of these compounds could be obtained by X-ray analysis. They exhibit interesting, but very different structural features: 1 forms chains that consist of molecules directly interconnected by N–H…N hydrogen bonds, whereas 2 retains the solvent molecule methanol in the “bay” region of the molecule to form stacked dimers. Structure 3 forms a chain-like structure, but retains the solvent molecule ethanol, which connects the molecules via hydrogen bonds. The idea was to synthesize europium(III) complexes containing the new ligands. Since the pyrrole amine is a very weak acid (pKa = 23.0) [17] it can only be deprotonated by hard bases, such as n-butyllithium or sodium hydride [18]. Therefore we chose to work in water-free conditions with the europium precursors EuCl3 (no crystallization water) or Eu[N(SiMe3)2]3, which are commercially available. To date we did not succeed in synthesizing the target complexes depicted on the right-hand side of Scheme 1. This class of compounds may also be of interest for other areas of chemistry. The pyrrole–pyridine structural motif is featured in current studies, owing to its complexation properties towards first-row transition metals (Fe, Co, Ni, Cu, Zn) [18] and ruthenium [19]. The intramolecular proton transfer of these species is also of interest for vibrational spectroscopy measurements [20].
Experimental
General
Melting points: Stuart Melting Point SMP3 apparatus, uncorr. Elemental analyses: Vario EL (Elementar Co.). IR: Bruker Tensor 27 spectrometer with a Diamond ATR sampling element. UV–vis: Varian Cary 100 Bio, spectra taken of solutions in spectroscopic grade solvents. NMR: 600 MHz (1H), 151 MHz (13C): Bruker AV2-600 spectrometer. 200 MHz (1H), 50 MHz (13C): Varian Mercury Plus 200. 1H chemical shifts were recorded with tetramethylsilane (TMS) as the internal standard. 13C measurements were taken with the corresponding solvent signal as the reference. J values are rounded to 0.1 Hz. Mass spectrometry: Thermofinnigan MAT95XL (EI). TLC: Silica plates (Polygram SIL G/UV 254), aluminium oxide plates (Polygram N/UV 254). Flash chromatography: Silica (Kieselgel 60, Fluka), aluminium oxide (aluminium oxide 90 neutral, Merck). Aluminium oxide, activity III, was made by adding 8% water and shaking the mixture vigorously in a closed flask. All reagents were purchased from Aldrich or Alfa Aesar and used as received. Solvents were purified before use. Dry solvents were purchased from Aldrich or Fluka. Reactions were performed under nitrogen atmosphere unless otherwise stated.
X-Ray structure determination: Data collection and reduction: Crystals were mounted in inert oil on glass fibres and transferred to the cold gas stream of the diffractometer (Oxford Diffraction Xcalibur). Measurements were performed with monochromated Mo Kα radiation (λ = 0.71073 Å). No absorption corrections were applied. Structure refinement: The structures were refined anisotropically against F2 (program SHELXL-97 [21]). Hydrogen atoms: OH and NH hydrogens were refined freely; methyl hydrogens as constituents of idealised rigid groups were allowed to rotate but not tip; other H were modelled by using a riding model starting from calculated positions. Exceptions and special features: Compound 1: in the absence of significant anomalous scattering, Friedel opposite reflections were merged and the Flack parameter is thus meaningless. Compound 3·C2H5OH: the ethanol molecule is disordered over two positions, but the minor component is occupied only to the extent of 9%. Its OH hydrogen was not located. Similarity restraints for both ethanol orientations were used to improve the stability of refinement. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications no. CCDC-871428 (1), CCDC-871429 (2·CH3OH), CCDC-871430 (3·C2H5OH). Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/data_request/cif.
Synthesis and characterization of the heteroaryl bromides 8 and 9 starting from 2,2’-bipyridine (11) and 1,10-phenanthroline (14), as well as the synthesis of compound 10 and 19 are shown in Supporting Information File 1. Absorption, excitation and emission spectra of compounds 1–3 are included as well.
Synthesis of 6-(1-tert-butoxycarbonylpyrrol-2-yl)-2,2’-bipyridine (4)
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-116-i13.svg?max-width=637&scale=1.18182)
Diisopropylamine (0.36 g, 3.57 mmol, 1.4 equiv) was dissolved in THF (5 mL) and cooled to −80 °C. Whilst the temperature was kept constant, n-butyllithium (2.25 mL, 1.6 M in hexane, 3.57 mmol, 1.4 equiv) was added dropwise, and the mixture was stirred for 1 h. Compound 19 (0.55 g, 3.29 mmol, 1.29 equiv) in THF (4 mL) was added dropwise at −80 °C, and the reaction mixture was stirred for another 1 h after which the reaction was quenched with trimethylborate (0.50 g, 4.85 mmol, 1.9 equiv). The mixture was allowed to warm to 0 °C and was stirred for 1.5 h. At room temperature 8 (0.6 g, 2.55 mmol) and Pd(PPh3)2Cl2 (144 mg, 0.2 mmol, 8 mol %) were added. The reaction mixture was heated under reflux and aq K2CO3 (5.1 mL, 1 M, 5.1 mmol, 2 equiv) was added meanwhile. It was kept at that temperature for 2 h, then cooled to room temperature and diluted with diethyl ether. The organic phase was washed with sat. aq NaCl and the aqueous phase was extracted two times with diethyl ether. The combined organic extracts were then dried over MgSO4, filtered and the solvent was removed. The raw product was preadsorbed onto silica. Column chromatography (SiO2, hexane/ethyl acetate 3:1, Rf = 0.4) gave 4 as a yellow resin (0.73 g, 2.27 mmol, 89%). 1H NMR (600 MHz, CDCl3) δ 8.67 (ddd, 3JH,H= 4.8 Hz, 4JH,H = 1.8 Hz, 5JH,H = 0.9 Hz, 1H, 1-H), 8.46 (pseudo-dt, 3JH,H = 8.0 Hz, J = 1.1 Hz, 1H, 4-H), 8.34 (dd, 3JH,H= 7.9 Hz, 4JH,H = 1.0 Hz, 1H, 7-H), 7.81 (dd, 3JH,H = 7.9 Hz, 3JH,H = 7.7 Hz, 1H, 8-H), 7.76 (ddd, 3JH,H = 7.9 Hz, 3JH,H = 7.6 Hz, 4JH,H = 1.8 Hz, 1H, 3-H), 7.43 (dd, 3JH,H= 7.7 Hz, 4JH,H = 1.0 Hz, 1H, 9-H), 7.41 (dd, 3JH,H= 3.3 Hz, 4JH,H = 1.8 Hz, 1H, 14-H), 7.28 (ddd, 3JH,H= 7.2 Hz, 3JH,H = 4.6 Hz, 4JH,H = 1.1 Hz, 1H, 2-H), 6.48 (dd, 3JH,H= 3.3 Hz, 4JH,H = 1.8 Hz, 1H, 12-H), 6.27 (dd, 3JH,H = 3.3 Hz, 3JH,H = 3.3 Hz, 1H, 13-H), 1.28 (s, 9H, 17-H) ppm; 13C NMR (151 MHz, CDCl3) δ 156.0 (s, C-5), 155.0 (s, C-6), 152.1 (s, C-10), 149.4 (s, C-15), 149.0 (d, C-1), 136.7 (d, C-8), 136.7 (d, C-3), 134.3 (s, C-11), 123.6 (d, C-2), 123.6 (d, C-14), 123.1 (d, C-9), 121.3 (d, C-4), 118.8 (d, C-7), 115.6 (d, C-12), 110.5 (d, C-13), 83.5 (s, C-16), 27.4 (q, C-17) ppm; EIMS (70 eV) m/z (% relative intensity): M+● 321 (5), [M − Boc]+● 222/221/220 (14/100/9); anal. calcd for C19H19N3O2: C 71.01, H 5.96, N 13.08; found: C 70.95, H 6.43, N 12.96.
Synthesis of 6-(pyrrol-2-yl)-2,2’-bipyridine (1)
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-116-i14.svg?max-width=637&scale=1.18182)
Compound 4 (1.5 g, 4.7 mmol) was dissolved in dichloromethane (110 mL) and cooled to 0 °C. Aqueous hydrochloric acid (27.5 mL, 2 M) was added dropwise under vigorous stirring, upon which the biphasic mixture turned yellow. The organic phase was separated, and the aqueous phase was neutralized with sodium carbonate and then extracted three times with dichloromethane. The combined organic extracts were dried over MgSO4 and filtered, and the solution was concentrated. Ethyl acetate (same volume as remaining dichloromethane) was added and the solution was filtered through aluminium oxide. Removal of the solvent yielded 1 as a colorless powder (0.96 g, 4.34 mmol, 93%). Crystals of 1 could be grown by recrystallization from ethanol. Mp 120–121.5 °C; 1H NMR (600 MHz, CDCl3) δ 8.68 (ddd, 3JH,H = 4.8 Hz, 4JH,H = 1.8 Hz, 5JH,H = 0.9 Hz, 1H, 1-H), 8.45 (pseudo-dt, 3JH,H = 8.0 Hz, J = 1.1 Hz, 1H, 4-H), 8.15 (dd, 3JH,H= 7.7 Hz, 4JH,H = 1.0 Hz, 1H, 7-H), 7.80 (ddd, 3JH,H = 7.9 Hz, 3JH,H = 7.5 Hz, 4JH,H = 1.8 Hz, 1H, 3-H), 7.75 (dd, 3JH,H= 7.9 Hz, 3JH,H = 7.7 Hz, 1H, 8-H), 7.56 (dd, 3JH,H= 7.9 Hz, 4JH,H = 1.0 Hz, 1H, 9-H), 7.30 (ddd, 3JH,H = 7.5 Hz, 3JH,H = 4.8 Hz, 4JH,H = 1.2 Hz, 1H, 2-H), 6.94 (pseudo-dt, J = 2.6 Hz, 4JH,H = 1.4 Hz, 1H, 14-H), 6.76 (ddd, 3JH,H = 3.7 Hz, J = 2.5 Hz, 4JH,H = 1.4 Hz, 1H, 12-H), 6.32 (pseudo-dt, 3JH,H = 3.6 Hz, J = 2.7 Hz, 1H, 13-H), 9.75 (br. s, 1H, N-H) ppm; 13C NMR (151 MHz, CDCl3) δ 156.2 (s, C-5), 155.0 (s, C-6), 149.9 (s, C-10), 149.1 (d, C-1), 137.4 (d, C-8), 136.7 (d, C-3), 131.6 (s, C-11), 123.6 (d, C-2), 121.0 (d, C-4), 119.7 (d, C-14), 118.2 (d, C-9), 117.9 (d, C-7), 110.3 (d, C-13), 107.3 (d, C-12) ppm; EIMS (70 eV) m/z (% relative intensity): M+● 222/221/220 (18/100/16); UV–vis (CH2Cl) λmax, nm (log ε): 307 (4.26), 283 (4.24), 239 (4.23); UV–vis (CH3OH) λmax, nm (log ε): 309 (4.29), 283 (4.22), 237 (4.23); IR (ATR) ![[Graphic 12]](/bjoc/content/inline/1860-5397-8-116-i18.svg?max-width=637&scale=1.18182) : 3126 (m), 3071 (m), 3005 (m), 2970 (m), 2841 (m), 2685 (m), 2551 (m), 1582 (m), 1555 (s), 1455 (s), 1428 (s), 1407 (m), 1328 (w), 1258 (m), 1159 (m), 1126 (s), 1098 (w), 1076 (w), 1061(w), 1033 (m), 1001 (w), 986 (w), 936 (w), 879 (m), 854 (m), 823 (m), 780 (s), 731 (s), 679 (m), 628 (m), 609 (m) cm−1; anal. calcd C14H11N3: C 76.00 , H 5.01, N 18.99; found: C 76.20, H 4.75, N 19.18.
: 3126 (m), 3071 (m), 3005 (m), 2970 (m), 2841 (m), 2685 (m), 2551 (m), 1582 (m), 1555 (s), 1455 (s), 1428 (s), 1407 (m), 1328 (w), 1258 (m), 1159 (m), 1126 (s), 1098 (w), 1076 (w), 1061(w), 1033 (m), 1001 (w), 986 (w), 936 (w), 879 (m), 854 (m), 823 (m), 780 (s), 731 (s), 679 (m), 628 (m), 609 (m) cm−1; anal. calcd C14H11N3: C 76.00 , H 5.01, N 18.99; found: C 76.20, H 4.75, N 19.18.
Synthesis of 2-(pyrrol-2-yl)-1,10-phenanthroline (2)
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-116-i15.svg?max-width=637&scale=1.18182)
Diisopropylamine (1.11 g, 11.0 mmol, 1.1 equiv) was dissolved in THF (20 mL) and cooled to −80 °C. Whilst the temperature was kept constant, n-butyllithium (7.5 mL, 1.6 M in hexane, 12.0 mmol, 1.2 equiv) was added dropwise, and the mixture was stirred for 1 h. Compound 19 (1.67 g, 10.0 mmol) in THF (5 mL) was added dropwise at −80 °C and the reaction mixture was stirred for another 1 h, after which it was quenched with trimethylborate (1.56 g, 15.0 mmol, 1.5 equiv). The mixture was allowed to warm to 0 °C and was stirred for 1.5 h. At room temperature 9 (2.85 g, 11.0 mmol, 1.1 equiv), Pd(PPh3)2Cl2 (555 mg, 0.8 mmol, 8 mol %) and THF (50 mL) were added. The reaction mixture was heated under reflux and aq K2CO3 (16.5 mL, 1 M, 16.5 mmol, 1.65 equiv) was added meanwhile. It was kept under reflux for 40 h, then cooled to room temperature and diluted with diethyl ether. The organic phase was washed with sat. aq NaCl, and the aqueous phase was extracted two times with diethyl ether. The combined organic extracts were then dried over MgSO4 and filtered, and the solvent was removed. The raw product was preadsorbed onto aluminium oxide, activity III. Column chromatography (Al2O3, activity III, dichloromethane/ethyl acetate/hexane 1:1:2 → dichloromethane/ethyl acetate 1:1, Rf (Al2O3, dichloromethane/ethyl acetate/hexane 1:1:2) = 0.27) gave 2 as a pale yellow solid (1.0 g, 4.1 mmol, 41%). Recrystallization from methanol gave brown crystals of 2, which retained one solvent molecule as indicated by X-ray and elemental analysis. Mp 119 °C (release of methanol), 137 °C (melting of the remaining solid); 1H NMR (600 MHz, CDCl3) δ 12.12 (s, 1H, N-H), 8.98 (dd, 3JH,H = 4.4 Hz, 4JH,H = 1.7 Hz, 1H, 1-H), 8.22 (dd, 3JH,H= 8.1 Hz, 4JH,H = 1.6 Hz, 1H, 3-H), 8.10 (d, 3JH,H = 8.5 Hz, 1H, 8-H), 7.88 (d, 3JH,H= 8.5 Hz, 1H, 9-H), 7.71 (d, 3JH,H= 8.7 Hz, 1H, 6-H), 7.64 (d, 3JH,H= 8.8 Hz, 1H, 5-H), 7.56 (dd, 3JH,H= 8.0 Hz, 3JH,H = 4.4 Hz, 1H, 2-H), 7.04 (dt, 3JH,H= 2.6 Hz, 4JH,H = 1.4 Hz, 1H, 14-H), 6.90 (ddd, 3JH,H = 3.7 Hz, J = 2.4 Hz, 4JH,H = 1.4 Hz, 1H, 15-H), 6.32 (pseudo-dt, 3JH,H= 3.6 Hz, J = 2.5 Hz, 1H, 16-H) ppm; 13C NMR (151 MHz, CDCl3) δ 150.9 (s, C-10), 148.8 (d, C-1), 145.5 (s, C-11), 145.2 (s, C-12), 136.6 (d, C-3), 136.2 (d, C-8), 132.3 (s, C-13), 129.1 (s, C-4), 126.8 (d, C-6), 126.4 (s, C-7), 124.4 (d, C-5), 122.8 (d, C-2), 122.0 (d, C-14), 119.1 (d, C-9), 109.8 (d, C-16), 109.2 (d, C-15) ppm; EIMS (70 eV) m/z (% relative intensity): [M]+● 247/246/245/244/243 (2/16/100/12/4); UV–vis (CH2Cl2) λmax, nm (log εmax): 339 (4.24), 311 (4.31), 235 (3.46); UV–vis (CH3OH) λmax, nm (log εmax): 345 (4.13), 312 (4.24), 237 (4.32) IR (ATR) :3610 (w), 3185 (m), 3113 (m), 2927 (w), 2820 (w), 1617 (w), 1585 (m), 1554 (m), 1503 (m), 1459 (m), 1423 (m), 1408 (m), 1378 (m), 1338 (w), 1262 (w), 1215 (w), 1145 (m), 1125 (s), 1080 (w), 1030 (s), 936 (w), 882 (w), 839 (s), 779 (s), 728 (s), 679 (s), 626 (m), 606 (s), 569 (m) cm−1; anal. calcd. for C16H11N3·CH3OH: C 73.63, H 5.45, N 15.15; found: C 73.30, H 5.33, N 15.25.
Synthesis of 2-(N-methylbenz[d,e]imidazo-2-yl)-6-(1-tert-butoxycarbonylpyrrol-2-yl)-pyridine (6)
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-116-i16.svg?max-width=637&scale=1.18182)
Diisopropylamine (0.22 g, 2.17 mmol, 1.3 equiv) was dissolved in THF (3 mL) and cooled to −80 °C. Whilst the temperature was kept constant, n-butyllithium (0.9 mL, 2.5 M in hexane, 2.25 mmol, 1.3 equiv) was added dropwise and the mixture was stirred for 1 h. Compound 19 (0.38 g, 2.27 mmol, 1.3 equiv) in THF (2 mL) was added dropwise at −80 °C and the reaction mixture was stirred for another 1 h, after which it was quenched with trimethylborate (0.23 g, 2.24 mmol, 1.3 equiv). The mixture was allowed to warm to 0 °C and was stirred for 0.5 h. At room temperature 10 (0.49 g, 1.70 mmol), Pd(PPh3)2Cl2 (0.07 g, 0.1 mmol, 6 mol %) and THF (4 mL) were added. The reaction mixture was heated under reflux, and aq K2CO3 (2.8 mL, 1 M, 2.8 mmol, 1.65 equiv) was added meanwhile. It was kept under reflux for 70 h, then cooled to room temperature and diluted with diethyl ether. The organic phase was washed with sat. aq NaCl and the aqueous phase was extracted two times with diethyl ether. The combined organic extracts were then dried over MgSO4 and filtered, and the solvent was removed. Column chromatography (SiO2, diethyl ether, Rf = 0.55) gave 6 as a colorless solid (0.55 g, 1.47 mmol, 85%). mp 125–126 °C; 1H NMR (600 MHz, CDCl3) δ 8.32 (dd, 3JH,H= 7.9 Hz, 4JH,H = 1.1 Hz, 1H, 11-H), 7.85 (dd, 3JH,H = 7.9 Hz, 3JH,H = 7.8 Hz, 1H, 10-H), 7.83 (ddd, 3JH,H= 7.4 Hz, 4JH,H = 1.6 Hz, 5JH,H = 0.7 Hz, 1H, 19-H), 7.46 (dd, 3JH,H= 7.8 Hz, 4JH,H = 1.0 Hz, 1H, 9-H), 7.43–7.41 (m, 1H, 16-H), 7.39 (dd, 3JH,H= 3.3 Hz, 4JH,H = 1.8 Hz, 1H, 4-H), 7.33 (ddd, 3JH,H = 7.5 Hz, 3JH,H = 7.1 Hz, 4JH,H = 1.4 Hz, 1H, 17-H), 7.30 (ddd, 3JH,H = 7.4 Hz, 3JH,H = 7.1 Hz, 4JH,H = 1.3 Hz), 6.50 (dd, 3JH,H = 3.3 Hz, 4JH,H = 1.7 Hz, 1H, 6-H), 6.28 (dd, 3JH,H = 3.3 Hz, 3JH,H = 3.3 Hz, 1H, 5-H), 4.25 (s, 3H, 14-H), 1.33 (s, 9H, 1-H) ppm; 13C NMR (151 MHz, CDCl3) δ 151.6 (s, C-8), 150.1 (s, C-13), 149.7 (s, C-12), 149.3 (s, C-3), 142.5 (s, C-20), 137.2 (s, C-15), 136.8 (d, C-10), 134.0 (s, C-7), 123.9 (d, C-4), 123.2 (d, C-17), 123.1 (d, C-9), 122.6 (d, C-11), 122.5 (d, C-18), 120.0 (d, C-19), 116.0 (d, C-6), 110.6 (d, C-5), 109.9 (d, C-16), 83.9 (s, C-2), 32.8 (q, C-14), 27.5 (q, C-1) ppm; EIMS (70 eV) m/z (% relative intensity): [M]+● 375/374 (2/6), [M – Boc + methyl]+● 290/289/288/287/286 (10/65/100/67/93), [M − Boc]+● 275/274/273 (6/32/35), [M − Boc-pyrrole + H+]+● 209/208/207/206 (3/18/16/12), [N-methylbenzimidazole]+● 132/131/130/129 (4/39/2/5); anal. calcd for C22 H22 N4 O2: C 70.57, H 5.92, N 14.96; found: C 70.39, H 5.97,N 15.02.
Synthesis of 2-(N-methylbenz[d,e]imidazo-2-yl)-6-(pyrrol-2-yl)-pyridine (3)
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-116-i17.svg?max-width=637&scale=1.18182)
Compound 6 (0.43 g, 1.15 mmol) was dissolved in methanol (35 mL). Sodium methanolate (0.13 g, 2.35 mmol, 2.0 equiv) was added and the solution was stirred for 17 h under reflux. The solvent was removed and the residue was taken up in ethanol (10 mL) and heated again. The mother liquor was then cooled to 0 °C leading to crystallization. Colorless crystals of 3 were collected (0.220 g, 0.802 mmol, 70%). mp 184–187 °C; 1H NMR (600 MHz, CDCl3) δ 9.83 (s, 1H, N-H), 7.97 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.0 Hz, 1H, 8-H), 7.85 (m, 1H, 16-H), 7.72 (dd, 3JH,H = 8.0 Hz, 3JH,H = 7.7 Hz, 1H, 7-H), 7.54 (dd, 3JH,H= 8.0 Hz, 4JH,H = 1.0 Hz, 1H, 6-H), 7.39–7.31 (m, 3H, 13-H, 14-H, 15-H), 7.00 (ddd, 3JH,H= 2.6 Hz, J = 2.6 Hz, 4JH,H = 1.4 Hz, 1H, 3-H), 6.77 (ddd, 3JH,H= 3.7 Hz, J = 2.5 Hz, 4JH,H = 1.4 Hz, 1H, 1-H), 6.35 (ddd, 3JH,H= 3.6 Hz, 3JH,H = 2.6 Hz, J = 2.6 Hz, 1H, 2-H), 4.10 (s, 3H, 11-H) ppm; 13C NMR (151 MHz, CDCl3) δ 150.8 (s, C-10), 149.7 (s, C-5), 149.3 (s, C-9), 142.5 (s, C-17), 137.3 (d, C-7), 136.9 (s, C-12), 131.4 (s, C-4), 123.2 (d, C-14), 122.6 (d, C-15), 121.7 (d, C-8), 120.2 (d, C-3), 120.0 (d, C-16), 118.2 (d, C-6), 110.5 (d, C-2), 109.9 (C-1), 107.8 (d, C-13), 32.4 (q, C-11) ppm; EIMS (70 eV) m/z (% relative intensity): [M]+● 276/275/274/273/272 (2/14/80/100/2); UV–vis (CH2Cl2) λmax, nm (log εmax): 335 (sh 4.20), 301 (4.41), 230 (4.22); UV–vis (CH3OH) λmax, nm (log εmax): 335 (sh 4.11), 300 (4.41), 232 (4.20); IR (ATR) : 3170 (m), 3102(m), 2965 (m), 2903 (m), 2858 (m), 1591 (m), 1566 (s), 1473 (s), 1454 (s), 1435 (s), 1391 (m), 1373 (m), 1327 (m), 1250 (m), 1158 (m), 1122 (m), 1083 (m), 1035 (m), 990 (m), 940 (w), 879 (m), 836 (m), 812 (s), 745 (s), 724 (s), 653 (m), 607 (m), 586 (w), 543 (m) cm−1; anal. calcd for C17H14N4: C 74.43, H 5.14, N 20.42; found: C 74.31, H 4.86, N 20.33.
Supporting Information
| Supporting Information File 1: Experimental details for known compounds and spectral data for compounds 1–3. | ||
| Format: PDF | Size: 497.0 KB | Download |
References
-
Shavaleev, N. M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 6178–6191. doi:10.1021/ic9005136
Return to citation in text: [1] [2] -
Shavaleev, N. M.; Gumy, F.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 5611–5613. doi:10.1021/ic9006372
Return to citation in text: [1] [2] [3] -
Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Chem.–Eur. J. 2009, 15, 10790–10802. doi:10.1002/chem.200901996
Return to citation in text: [1] [2] [3] -
Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2010, 49, 3927–3936. doi:10.1021/ic100129r
Return to citation in text: [1] [2] [3] [4] -
Bortoluzzi, M.; Paolucci, G.; Polizzi, S.; Bellotto, L.; Enrichi, F.; Ciorba, S.; Richards, B. S. Inorg. Chem. Commun. 2011, 14, 1762–1766. doi:10.1016/j.inoche.2011.08.004
Return to citation in text: [1] -
Melby, L. R.; Rose, N. J.; Abramson, E.; Caris, J. C. J. Am. Chem. Soc. 1964, 86, 5117–5125. doi:10.1021/ja01077a015
Return to citation in text: [1] [2] [3] -
Bauer, H.; Blanc, J.; Ross, D. L. J. Am. Chem. Soc. 1964, 5125–5131. doi:10.1021/ja01077a016
Return to citation in text: [1] -
Johannes, H.-H.; Kowalsky, W.; Zoellner, M.; Böttger, M.; Wiegmann, B. Verfahren und Verwendung lumineszenter Verbindungen zur Messung einer photokatalytischen Oberflächenaktivität. DE102009043378B4, April 7, 2011.
Return to citation in text: [1] -
Filipescu, N.; Mushrush, G. W.; Hurt, C. R.; McAvoy, N. Nature 1966, 211, 960–961. doi:10.1038/211960a0
Return to citation in text: [1] -
Knapp, D. M.; Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963. doi:10.1021/ja901416p
Return to citation in text: [1] [2] -
Roche, A. J.; Canturk, B. Org. Biomol. Chem. 2005, 3, 515–519. doi:10.1039/b415764h
Return to citation in text: [1] [2] -
Norrby, T.; Börje, A.; Zhang, L.; Åkermark, B. Acta Chem. Scand. 1998, 52, 77–85. doi:10.3891/acta.chem.scand.52-0077
Return to citation in text: [1] [2] -
Halcrow, B. E.; Kermack, W. O. J. Chem. Soc. 1946, 155–157. doi:10.1039/jr9460000155
Return to citation in text: [1] -
Nair, S. K.; Planken, S. P.; Plewe, M. B.; Vernier, W. F.; Yang, Y.; Zhu, H. Benzimidazole Derivatives. WO2011/027249A3, March 10, 2011.
Return to citation in text: [1] -
Grehn, L.; Ragnarsson, U. Angew. Chem., Int. Ed. Engl. 1984, 23, 296–301. doi:10.1002/anie.198402961
Return to citation in text: [1] -
Allen, F. H. Acta Crystallogr., Sect. B 2002, 58, 380–388. doi:10.1107/S0108768102003890
Return to citation in text: [1] -
Bordwell, F. G.; Drucker, G. E.; Fried, H. E. J. Org. Chem. 1981, 46, 632–635. doi:10.1021/jo00316a032
Return to citation in text: [1] -
Ciszek, J. W.; Keane, Z. K.; Cheng, L.; Stewart, M. P.; Yu, L. H.; Natelson, D.; Tour, J. M. J. Am. Chem. Soc. 2006, 128, 3179–3189. doi:10.1021/ja055459d
Return to citation in text: [1] [2] -
Nagata, T.; Tanaka, K. Bull. Chem. Soc. Jpn. 2002, 75, 2469–2470. doi:10.1246/bcsj.75.2469
Return to citation in text: [1] -
Kijak, M.; Nosenko, Y.; Singh, A.; Thummel, R. P.; Brutschy, B.; Waluk, J. J. Mol. Struct. 2007, 844–845, 286–299. doi:10.1016/j.molstruc.2007.05.019
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1]
| 18. | Ciszek, J. W.; Keane, Z. K.; Cheng, L.; Stewart, M. P.; Yu, L. H.; Natelson, D.; Tour, J. M. J. Am. Chem. Soc. 2006, 128, 3179–3189. doi:10.1021/ja055459d |
| 16. | Allen, F. H. Acta Crystallogr., Sect. B 2002, 58, 380–388. doi:10.1107/S0108768102003890 |
| 17. | Bordwell, F. G.; Drucker, G. E.; Fried, H. E. J. Org. Chem. 1981, 46, 632–635. doi:10.1021/jo00316a032 |
| 1. | Shavaleev, N. M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 6178–6191. doi:10.1021/ic9005136 |
| 2. | Shavaleev, N. M.; Gumy, F.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 5611–5613. doi:10.1021/ic9006372 |
| 3. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Chem.–Eur. J. 2009, 15, 10790–10802. doi:10.1002/chem.200901996 |
| 4. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2010, 49, 3927–3936. doi:10.1021/ic100129r |
| 5. | Bortoluzzi, M.; Paolucci, G.; Polizzi, S.; Bellotto, L.; Enrichi, F.; Ciorba, S.; Richards, B. S. Inorg. Chem. Commun. 2011, 14, 1762–1766. doi:10.1016/j.inoche.2011.08.004 |
| 8. | Johannes, H.-H.; Kowalsky, W.; Zoellner, M.; Böttger, M.; Wiegmann, B. Verfahren und Verwendung lumineszenter Verbindungen zur Messung einer photokatalytischen Oberflächenaktivität. DE102009043378B4, April 7, 2011. |
| 15. | Grehn, L.; Ragnarsson, U. Angew. Chem., Int. Ed. Engl. 1984, 23, 296–301. doi:10.1002/anie.198402961 |
| 2. | Shavaleev, N. M.; Gumy, F.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 5611–5613. doi:10.1021/ic9006372 |
| 3. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Chem.–Eur. J. 2009, 15, 10790–10802. doi:10.1002/chem.200901996 |
| 4. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2010, 49, 3927–3936. doi:10.1021/ic100129r |
| 11. | Roche, A. J.; Canturk, B. Org. Biomol. Chem. 2005, 3, 515–519. doi:10.1039/b415764h |
| 6. | Melby, L. R.; Rose, N. J.; Abramson, E.; Caris, J. C. J. Am. Chem. Soc. 1964, 86, 5117–5125. doi:10.1021/ja01077a015 |
| 7. | Bauer, H.; Blanc, J.; Ross, D. L. J. Am. Chem. Soc. 1964, 5125–5131. doi:10.1021/ja01077a016 |
| 14. | Nair, S. K.; Planken, S. P.; Plewe, M. B.; Vernier, W. F.; Yang, Y.; Zhu, H. Benzimidazole Derivatives. WO2011/027249A3, March 10, 2011. |
| 6. | Melby, L. R.; Rose, N. J.; Abramson, E.; Caris, J. C. J. Am. Chem. Soc. 1964, 86, 5117–5125. doi:10.1021/ja01077a015 |
| 4. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2010, 49, 3927–3936. doi:10.1021/ic100129r |
| 10. | Knapp, D. M.; Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963. doi:10.1021/ja901416p |
| 12. | Norrby, T.; Börje, A.; Zhang, L.; Åkermark, B. Acta Chem. Scand. 1998, 52, 77–85. doi:10.3891/acta.chem.scand.52-0077 |
| 20. | Kijak, M.; Nosenko, Y.; Singh, A.; Thummel, R. P.; Brutschy, B.; Waluk, J. J. Mol. Struct. 2007, 844–845, 286–299. doi:10.1016/j.molstruc.2007.05.019 |
| 10. | Knapp, D. M.; Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963. doi:10.1021/ja901416p |
| 12. | Norrby, T.; Börje, A.; Zhang, L.; Åkermark, B. Acta Chem. Scand. 1998, 52, 77–85. doi:10.3891/acta.chem.scand.52-0077 |
| 13. | Halcrow, B. E.; Kermack, W. O. J. Chem. Soc. 1946, 155–157. doi:10.1039/jr9460000155 |
| 21. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 1. | Shavaleev, N. M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 6178–6191. doi:10.1021/ic9005136 |
| 2. | Shavaleev, N. M.; Gumy, F.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 5611–5613. doi:10.1021/ic9006372 |
| 3. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Chem.–Eur. J. 2009, 15, 10790–10802. doi:10.1002/chem.200901996 |
| 4. | Shavaleev, N. M.; Eliseeva, S. V.; Scopelliti, R.; Bünzli, J.-C. G. Inorg. Chem. 2010, 49, 3927–3936. doi:10.1021/ic100129r |
| 18. | Ciszek, J. W.; Keane, Z. K.; Cheng, L.; Stewart, M. P.; Yu, L. H.; Natelson, D.; Tour, J. M. J. Am. Chem. Soc. 2006, 128, 3179–3189. doi:10.1021/ja055459d |
| 6. | Melby, L. R.; Rose, N. J.; Abramson, E.; Caris, J. C. J. Am. Chem. Soc. 1964, 86, 5117–5125. doi:10.1021/ja01077a015 |
| 9. | Filipescu, N.; Mushrush, G. W.; Hurt, C. R.; McAvoy, N. Nature 1966, 211, 960–961. doi:10.1038/211960a0 |
| 11. | Roche, A. J.; Canturk, B. Org. Biomol. Chem. 2005, 3, 515–519. doi:10.1039/b415764h |
| 19. | Nagata, T.; Tanaka, K. Bull. Chem. Soc. Jpn. 2002, 75, 2469–2470. doi:10.1246/bcsj.75.2469 |
© 2012 Böttger et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)