Abstract
The palladium-catalyzed nucleophilic substitution of (coumarinyl)methyl acetates is described. The reaction proceeds though a palladium π-benzyl-like complex and allows for many different types of C-, N-, and S-nucleophiles to be regioselectively added to the biologically active coumarin motif. This new method was utilized to prepare a 128-membered library of aminated coumarins for biological screening.

Graphical Abstract
Introduction
Coumarins are privileged chemical motifs found in many natural products and drug molecules [1-15]. Because of their biological significance, there have been many classic and modern methods developed for the synthesis of these useful core structures [16-24]. Due to the ambiphilic nature of the heterocyclic ring of coumarin, this core-structure undergoes a diverse array of coupling reactions, such as halogenations [25], cycloadditions [26-32], conjugate additions [33-37] and transition-metal-catalyzed C–H activation/coupling reactions [38-44].
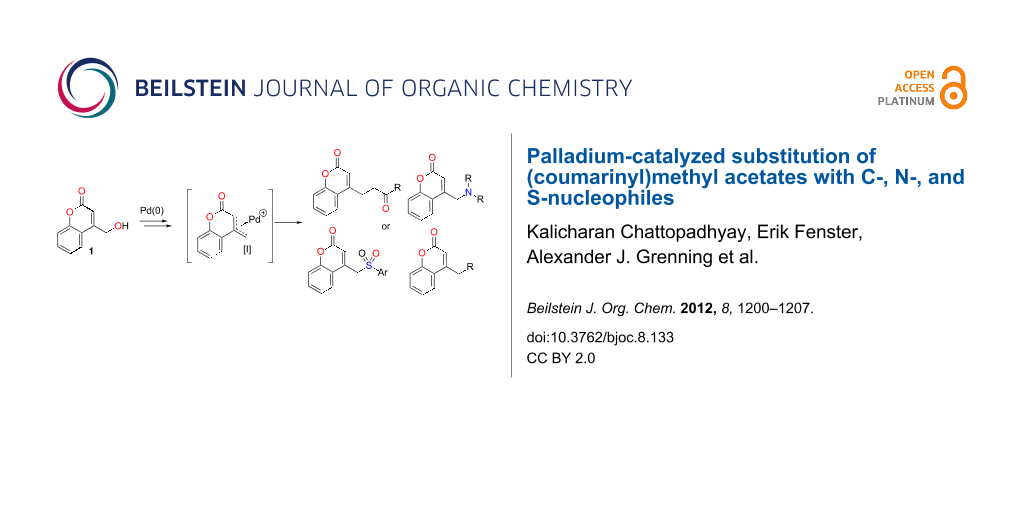
Substituted methylcoumarins, including aminomethylcoumarins are important biologically active motifs (Figure 1) [6-15]. Substitution of methylcoumarins to form compounds akin to 2 typically utilizes the corresponding halomethylcoumarin and highly stabilized nucleophiles or amines [6-15]. Due to the sensitivity and toxicity of related benzyl halides, there has been interest in catalytically activating hydroxymethylarene and heteroarenes (e.g., benzyl alcohol derivatives) toward reactions with nucleophiles [45-55]. In this realm, we [53,55] and others [54] have focused efforts on catalyzing benzylic substitutions with less-stabilized (DMSO pKa ~ 20–30) nucleophiles through decarboxylative coupling. In the present context, we hypothesized that a broad diversity of nucleophiles could be added to the (coumarinyl)methyl core through palladium-catalyzed couplings of hydroxymethylcoumarin derivatives. Herein we report that hydroxymethylcoumarin derivatives of type-1 undergo selective nucleophilic substitution at the exo-methyl position with C-, N-, and S-based nucleophiles by using palladium(0) as a catalyst (Scheme 1). In addition, given the known biological activity of aminomethylcoumarins, we prepared a 128-member library of aminated coumarins using rapid automated synthesis.
![[1860-5397-8-133-1]](/bjoc/content/figures/1860-5397-8-133-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative biologically active aminomethylcoumarins.
Figure 1: Representative biologically active aminomethylcoumarins.
![[1860-5397-8-133-i1]](/bjoc/content/inline/1860-5397-8-133-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Approach to diversely substituted coumarins.
Scheme 1: Approach to diversely substituted coumarins.
Results and Discussion
To begin investigating the diversification of hydroxymethylcoumarins, we chose to investigate their decarboxylative couplings of enolates. We have previously shown that decarboxylative benzylation (DcB) is a useful method for the addition of less-stabilized enolate anions to a benzyl functionality [46,53-55]. Thus, we envisioned being able to add various ketone enolates to the exo-methyl position of the coumarin core by this method (Table 1). Screening of reaction conditions showed that, in contrast to common decarboxylative benzylation conditions (Table 1,entries 1 and 2) under which nonpolar solvents give the best DcB, the selective mono-alkylation of the enolate proceeds in highest yield in acetonitrile (2a, Table 1, entry 3). Bidentate ligated palladium complexes gave mixed results: the Pd/dppf complex (Table 1,entry 4) catalyzed the reaction smoothly, while little product was seen on using the Pd/BINAP complex (Table 1,entry 5). Under the best conditions (5 mol % Pd(PPh3)4, MeCN, rt, Table 1, entry 3), a variety of enolate nucleophiles could be selectively coupled with coumarin electrophiles (Scheme 2). For example, acetone, acetophenone and 1,1,1-trimethylacetone enolates can all be generated and coupled with the coumarin electrophile. Importantly, selective monobenzylation was achieved for each example. Regarding the coumarin moiety, the decarboxylative coupling was compatible with a variety of simple substitutions, including methoxy (2c,d,g,h), chloro (2g,h) and naphthyl (2e) coumarins.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-133-i7.svg?max-width=637&scale=1.0)
![[1860-5397-8-133-i2]](/bjoc/content/inline/1860-5397-8-133-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the decarboxylative coupling.
Scheme 2: Scope of the decarboxylative coupling.
While the decarboxylative coupling worked well for the couplings of relatively nonstabilized enolate nucleophiles, we wished to find a more universal starting material that would allow a broader range of nucleophiles to be coupled to the coumarin core in an intermolecular fashion. Such a method was deemed necessary for rapid diversification and chemical library synthesis. Thus, we chose to investigate the Pd-catalyzed benzylic substitution reaction of (coumarinyl)methyl acetates with various nucleophiles. In the forthcoming sections, the coupling of this coumarin template to arylboronic acids [56-61], amines [47] and arylsulfinates is described [62]. Moreover, compared to related methods for palladium-catalyzed benzylic substitution that require specialized ligands [45,47,60-62], we report that coumarin π-benzyl formation is easily achieved with simple PPh3 ligated palladium.
Regarding the development of the Suzuki-like coupling reaction of (coumarinyl)methyl acetate and arylboronic acids, we screened reaction conditions for the coupling of coumarin 1a with phenylboronic acid (Table 2). We were pleased to find that the reaction progressed reasonably well to 3a under various conditions. For example, inorganic bases such as K2CO3, K3PO4, and KF all effected the reaction equally well in methanol. A brief screen of solvents showed that the nonpolar aprotic solvent 1,4-dioxane gave the highest yields of the coupling product (Table 2, entry 6). Lastly, in the absence of added base, the acetate generated upon π-benzyl formation also promoted the desired coupling, albeit in lower yield over the time frame allowed (12 h, Table 2, entry 7).
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-133-i8.svg?max-width=637&scale=1.0)
With the optimal conditions in hand, we next tested the scope of the reaction using various boronic acid and coumarinyl acetates (Scheme 3). Regarding the boronic acid coupling partner, modifications including fluoro and alkyl substitution were tolerated. In addition to aryl boronic acids (3a–c), the reaction was also extended to couplings of vinyl boronic acids having varied electronic properties (3d–e). As before, various simple substitutions and electronic changes were tolerated on the coumarin core.
![[1860-5397-8-133-i3]](/bjoc/content/inline/1860-5397-8-133-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of Suzuki-coupling of coumarinyl acetates.
Scheme 3: Scope of Suzuki-coupling of coumarinyl acetates.
In addition to the above Suzuki couplings, acetoxymethylcoumarins were found to undergo palladium-catalyzed substitution with sulfinates and secondary amines under the previously developed palladium-catalysis conditions (Scheme 4). Importantly, related aminomethylcoumarins have been shown to have significant biological activity [6-15].
![[1860-5397-8-133-i4]](/bjoc/content/inline/1860-5397-8-133-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Coupling of (coumarinyl)methyl acetates with N- and S-nucleophiles.
Scheme 4: Coupling of (coumarinyl)methyl acetates with N- and S-nucleophiles.
Regarding the coupling reaction between aryl sulfinates and the coumarinyl acetate, the reaction could be performed without the addition of an external base, since the aryl sulfinate is administered as its anion (Scheme 5). Phenyl (4a–c) and tolyl (4d–e) sulfinates were viable coupling partners, giving the product sulfones in good yield. As previously noted, the coumarin core tolerated various simple electronic (4b,c,e) and alkyl (4a,c,d) substitution patterns.
![[1860-5397-8-133-i5]](/bjoc/content/inline/1860-5397-8-133-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Scope of the coumarinyl acetate and aryl sulfinate coupling reaction.
Scheme 5: Scope of the coumarinyl acetate and aryl sulfinate coupling reaction.
Since (coumarinyl)methylamines are known to possess interesting biological activity, the scope of the amination was investigated in somewhat more detail. Regarding the scope of the amine and coumarinyl acetate coupling reaction, dialkyl (5a–c) and the cyclic amines pyrrolidine (5d–5f), pyrazine (5g), and morpholine (5h) were competent coupling partners (Scheme 6). Primary amines were also compatible coupling partners, although under these conditions, the reaction never went to full conversion (5i–k).
![[1860-5397-8-133-i6]](/bjoc/content/inline/1860-5397-8-133-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Scope of the coumarinyl acetate and amine coupling reaction.
Scheme 6: Scope of the coumarinyl acetate and amine coupling reaction.
As mentioned previously, we were devising this approach to coumarin substitution not only for the sake of new chemical methodology, but also because this approach could easily lead to rapid library synthesis. As aminomethylcoumarins are a biologically active chemical motif [6-15], we set out to produce a 128-member library of aminated coumarins using our method, with the goal of making unique, biologically active molecules (Figure 2). To begin, an 8 × 16 library (coumarin C1–8 × amine A1–16) of aminomethylcoumarins was devised for preparation by using a Chemspeed SLT100 automated synthesizer. For automated synthesis, the procedure and workflow was modified somewhat for optimal yield. Specifically, the loading of Pd was lowered to 3 mol % and the quantity of amine was raised to 1.5 equivalents to ensure complete conversion. Using the workflow shown in Figure 2 for the reaction of coumarin C3 with diallylamine A4, followed by filtration through a silica SPE, produced product C3A4 in 74% yield in a test case. In addition, the product was determined to contain 2% PPh3 impurity. Thus, it was concluded that the entire library should be purified by mass-directed fractionation to ensure high-quality compounds for biological screening.
![[1860-5397-8-133-2]](/bjoc/content/figures/1860-5397-8-133-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Library planning for amine (A) and coumarin (C) coupling partners.
Figure 2: Library planning for amine (A) and coumarin (C) coupling partners.
Regarding the library, coumarins and amines were chosen with diverse structures and properties in order to access the most chemical space within the aminomethylcoumarin family (Figure 2). The coumarins utilized had various electronic properties and substitution patterns. A polyaromatic coumarin was also screened (C4). Similarly, a diverse array of amines was chosen. Aside from simple dialkylamines, cyclic and heteroatom-containing amines were utilized. Amines with various functional groups, such as amides, aromatics, olefins, and alkyne substitution patterns, were also incorporated in this library. The automated library synthesis had a success rate of >85%, with the desired products isolated in high purity [63]. Taking center cuts by using mass-directed fractionation ensured that compounds were isolated in >95% purity; however, purity was achieved at some expense to the isolated yields, which were typically lower (5–75%) than those achieved in batch reactions. While most compounds were obtained in sufficient quantity for biological screening, the yields were variable (Figure 3). In particular, coumarins C6, C2 and C8 provided lower average yields than the other coumarin cores. Thus, it appears that 6,7-substitution of the coumarin core is somewhat problematic. In addition, analysis of the library yields indicates that N-arylpiperazines A8 and A9 in addition to propargylamine A15 were the most problematic. Perhaps unsurprisingly, the amine that provided the highest yields and highest success rate was diallyl amine, which was used for initial validation of the Chemspeed method (see above). In addition, piperidine and the simple open-chain dialkyl amines also provided good yields and high success rates.
![[1860-5397-8-133-3]](/bjoc/content/figures/1860-5397-8-133-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Results for the synthesis of a 128-member library of aminated coumarins by using the Chemspeed SLT100 automated synthesizer. Purified by an automated preparative reverse-phase HPLC (Waters 2767 Mass Directed Fractionation) detected by UV (270 nm). Purity was determined by reverse-phase HPLC (Waters Alliance 2795 system) with peak area (UV) at 214 nm.
Figure 3: Results for the synthesis of a 128-member library of aminated coumarins by using the Chemspeed SLT1...
Conclusion
In conclusion, we have developed a simple and general strategy for the palladium-catalyzed substitution of coumarins selectively at an exomethyl position. This approach allowed the coupling of various C-,N-, and S-based nucleophiles under mild conditions. We also utilized this approach in the synthesis of a 128-member chemical library appropriate for biological screening.
Supporting Information
| Supporting Information File 1: Experimental data and 1H, 13C, and IR, and HRMS data for new compounds produced in batch. | ||
| Format: PDF | Size: 3.7 MB | Download |
| Supporting Information File 2: Detailed results of the library analysis are likewise included. | ||
| Format: PDF | Size: 233.5 KB | Download |
References
-
Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriate, E. Curr. Med. Chem. 2005, 12, 887–916. doi:10.2174/0929867053507315
Return to citation in text: [1] -
Estévez-Braun, A.; González, A. G. Nat. Prod. Rep. 1997, 14, 465–475. doi:10.1039/np9971400465
Return to citation in text: [1] -
O'Kennedy, R.; Thornes, R. D., Eds. Coumarins: Biology, Applications and Mode of Action; John Wiley & Sons: Chichester, U.K., 1997.
Return to citation in text: [1] -
Forsyth, C. J.; Ying, L.; Chen, J.; La Clair, J. J. J. Am. Chem. Soc. 2006, 128, 3858–3859. doi:10.1021/ja057087e
Return to citation in text: [1] -
Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Bioorg. Med. Chem. Lett. 2008, 18, 423–426. doi:10.1016/j.bmcl.2007.09.100
Return to citation in text: [1] -
Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u
Return to citation in text: [1] [2] [3] [4] [5] -
Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111
Return to citation in text: [1] [2] [3] [4] [5] -
Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435
Return to citation in text: [1] [2] [3] [4] [5] -
Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127
Return to citation in text: [1] [2] [3] [4] [5] -
Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a
Return to citation in text: [1] [2] [3] [4] [5] -
Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007
Return to citation in text: [1] [2] [3] [4] [5] -
Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y
Return to citation in text: [1] [2] [3] [4] [5] -
Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149
Return to citation in text: [1] [2] [3] [4] [5] -
Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j
Return to citation in text: [1] [2] [3] [4] [5] -
Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q
Return to citation in text: [1] [2] [3] [4] [5] -
Perkin, W. H. J. Chem. Soc. 1868, 21, 53–63. doi:10.1039/JS8682100053
Return to citation in text: [1] -
Woodruff, E. H. Org. Synth. 1955, Coll. Vol. 3, 581.
Return to citation in text: [1] -
Kelkar, R. M.; Joshi, U. K.; Paradkar, M. V. Synthesis 1986, 214–216. doi:10.1055/s-1986-31620
Return to citation in text: [1] -
Armstrong, V.; Soto, O.; Valderrama, J. A.; Tapia, R. Synth. Commun. 1988, 18, 717–725. doi:10.1080/00397918808077361
Return to citation in text: [1] -
Darvatkar, N. B.; Deorukhkar, A. R.; Bhilare, S. V.; Raut, D. G.; Salunkhe, M. M. Synth. Commun. 2008, 38, 3508–3513. doi:10.1080/00397910802162967
Return to citation in text: [1] -
Sato, K.; Amakasu, T.; Abe, S. J. Org. Chem. 1964, 29, 2971–2972. doi:10.1021/jo01033a040
Return to citation in text: [1] -
Hepworth, J. D. In Comprehensive Heterocyclic Chemistry; Katritzky, A.; Rees, C. W.; Boulton, A. J.; McKillop, A., Eds.; Pergamon: Oxford, U.K., 1984; Vol. 3, pp 799–810.
Return to citation in text: [1] -
Zeitler, K.; Rose, C. A. J. Org. Chem. 2009, 74, 1759–1762. doi:10.1021/jo802285r
Return to citation in text: [1] -
Ramachandra, M. S.; Subbaraju, G. V. Synth. Commun. 2006, 36, 3723–3727. doi:10.1080/10916460600946154
Return to citation in text: [1] -
Kim, K.-M.; Park, I.-H. Synthesis 2004, 2004, 2641–2644. doi:10.1055/s-2004-831232
Return to citation in text: [1] -
Ballerini, E.; Minuti, L.; Piermatti, O.; Pizzo, F. J. Org. Chem. 2009, 74, 4311–4317. doi:10.1021/jo9005365
Return to citation in text: [1] -
Richardson, T. I.; Dodge, J. A.; Durst, G. L.; Pfeifer, L. A.; Shah, J.; Wang, Y.; Durbin, J. D.; Krishnan, V.; Norman, B. H. Bioorg. Med. Chem. Lett. 2007, 17, 4824–4828. doi:10.1016/j.bmcl.2007.06.052
Return to citation in text: [1] -
Girotti, R.; Marrocchi, A.; Minuti, L.; Piermatti, O.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2006, 71, 70–74. doi:10.1021/jo051539o
Return to citation in text: [1] -
Haight, A. R.; Bailey, A. E.; Baker, W. S.; Cain, M. H.; Copp, R. R.; DeMattei, J. A.; Ford, K. L.; Henry, R. F.; Hsu, M. C.; Keyes, R. F.; King, S. A.; McLaughlin, M. A.; Melcher, L. M.; Nadler, W. R.; Oliver, P. A.; Parekh, S. I.; Patel, H. H.; Seif, L. S.; Staeger, M. A.; Wayne, G. S.; Wittenberger, S. J.; Zhang, W. Org. Process Res. Dev. 2004, 8, 897–902. doi:10.1021/op049889k
Return to citation in text: [1] -
Ohkata, K.; Lee, Y. G.; Utsumi, Y.; Ishimaru, K.; Akiba, K. J. Org. Chem. 1991, 56, 5052–5059. doi:10.1021/jo00017a014
Return to citation in text: [1] -
Yamago, S.; Nakamura, E. J. Am. Chem. Soc. 1989, 111, 7285–7286. doi:10.1021/ja00200a072
Return to citation in text: [1] -
Trost, B. M.; Chan, D. M. T. J. Am. Chem. Soc. 1981, 103, 5972–5974. doi:10.1021/ja00409a083
Return to citation in text: [1] -
Teichert, J. F.; Feringa, B. L. Chem. Commun. 2011, 47, 2679–2681. doi:10.1039/c0cc05160h
Return to citation in text: [1] -
Huang, X.; Wen, Y.-H.; Zhou, F.-T.; Chen, C.; Xu, D.-C.; Xie, J.-W. Tetrahedron Lett. 2010, 51, 6637–6640. doi:10.1016/j.tetlet.2010.10.055
Return to citation in text: [1] -
Wang, C.; Tunge, J. A. Org. Lett. 2005, 7, 2137–2139. doi:10.1021/ol050466s
Return to citation in text: [1] -
Chen, G.; Tokunaga, N.; Hayashi, T. Org. Lett. 2005, 7, 2285–2288. doi:10.1021/ol0507367
Return to citation in text: [1] -
Dixon, D. J.; Ley, S. V.; Rodríguez, F. Angew. Chem., Int. Ed. 2001, 40, 4763–4765. doi:10.1002/1521-3773(20011217)40:24<4763::AID-ANIE4763>3.0.CO;2-D
Return to citation in text: [1] -
Wunderlich, S. H.; Rohbogner, C. J.; Unsinn, A.; Knochel, P. Org. Process Res. Dev. 2010, 14, 339–345. doi:10.1021/op9002888
Return to citation in text: [1] -
Martins, S.; Branco, P. S.; de la Torre, M. C.; Sierra, M. A.; Pereira, A. Synlett 2010, 2918–2922. doi:10.1055/s-0030-1259014
Return to citation in text: [1] -
Jeganmohan, M.; Knochel, P. Angew. Chem., Int. Ed. 2010, 49, 8520–8524. doi:10.1002/anie.201003558
Return to citation in text: [1] -
Huang, B.; Liu, J.; Huang, W. J. Chem. Soc., Chem. Commun. 1990, 1781–1782. doi:10.1039/C39900001781
Return to citation in text: [1] -
Jana, R.; Partridge, J. J.; Tunge, J. A. Angew. Chem., Int. Ed. 2011, 50, 5157–5161. doi:10.1002/anie.201100765
Return to citation in text: [1] -
Jana, R.; Trivedi, R.; Tunge, J. A. Org. Lett. 2009, 11, 3434–3436. doi:10.1021/ol901288r
Return to citation in text: [1] -
Chattopadhyay, K.; Jana, R.; Day, V. W.; Douglas, J. T.; Tunge, J. A. Org. Lett. 2010, 12, 3042–3045. doi:10.1021/ol101042x
Return to citation in text: [1] -
Kuwano, R. Synthesis 2009, 2009, 1049–1061. doi:10.1055/s-0028-1088001
Return to citation in text: [1] [2] -
Weaver, J. D.; Recio, A., III; Grenning, A. J.; Tunge, J. A. Chem. Rev. 2011, 111, 1846–1913. doi:10.1021/cr1002744
Return to citation in text: [1] [2] -
Kuwano, R.; Kondo, Y.; Matsuyama, Y. J. Am. Chem. Soc. 2003, 125, 12104–12105. doi:10.1021/ja037735z
Return to citation in text: [1] [2] [3] -
Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2012, 134, 5778–5781. doi:10.1021/ja301461p
Return to citation in text: [1] -
Zhu, Y.; Rawal, V. H. J. Am. Chem. Soc. 2011, 134, 111–114. doi:10.1021/ja2095393
Return to citation in text: [1] -
Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2010, 12, 1360–1363. doi:10.1021/ol1002576
Return to citation in text: [1] -
Legros, J.-Y.; Toffano, M.; Fiaud, J.-C. Tetrahedron 1995, 51, 3235–3246. doi:10.1016/0040-4020(95)00061-C
Return to citation in text: [1] -
Legros, J.-Y.; Fiaud, J.-C. Tetrahedron Lett. 1992, 33, 2509–2510. doi:10.1016/S0040-4039(00)92227-5
Return to citation in text: [1] -
Torregrosa, R. R. P.; Ariyarathna, Y.; Chattopadhyay, K.; Tunge, J. A. J. Am. Chem. Soc. 2010, 132, 9280–9282. doi:10.1021/ja1035557
Return to citation in text: [1] [2] [3] -
Fields, W. H.; Chruma, J. J. Org. Lett. 2010, 12, 316–319. doi:10.1021/ol902651j
Return to citation in text: [1] [2] [3] -
Recio, A., III; Heinzman, J. D.; Tunge, J. A. Chem. Commun. 2012, 48, 142–144. doi:10.1039/c1cc16011g
Return to citation in text: [1] [2] [3] -
Chowdhury, S.; Georghiou, P. E. Tetrahedron Lett. 1999, 40, 7599–7603. doi:10.1016/S0040-4039(99)01566-X
Return to citation in text: [1] -
Chahen, L.; Doucet, H.; Santelli, M. Synlett 2003, 1668–1672. doi:10.1055/s-2003-40994
Return to citation in text: [1] -
Nobre, S. M.; Monteiro, A. L. Tetrahedron Lett. 2004, 45, 8225–8228. doi:10.1016/j.tetlet.2004.09.020
Return to citation in text: [1] -
Yen, S. K.; Koh, L. L.; Huynh, H. V.; Hor, T. S. A. Eur. J. Inorg. Chem. 2009, 4288–4297. doi:10.1002/ejic.200900447
Return to citation in text: [1] -
Kuwano, R.; Yokogi, M. Chem. Commun. 2005, 5899–5901. doi:10.1039/B513372F
Return to citation in text: [1] [2] -
Kuwano, R.; Yokogi, M. Org. Lett. 2005, 7, 945–947. doi:10.1021/ol050078q
Return to citation in text: [1] [2] -
Kuwano, R.; Kondo, Y.; Shirahama, T. Org. Lett. 2005, 7, 2973–2975. doi:10.1021/ol0509787
See for related coupling of benzyl acetates with sulfinates.
Return to citation in text: [1] [2] -
“Success rate” is defined as >10 mg isolated in >95% purity.
Return to citation in text: [1]
| 6. | Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u |
| 7. | Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111 |
| 8. | Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435 |
| 9. | Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127 |
| 10. | Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a |
| 11. | Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007 |
| 12. | Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y |
| 13. | Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149 |
| 14. | Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j |
| 15. | Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q |
| 6. | Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u |
| 7. | Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111 |
| 8. | Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435 |
| 9. | Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127 |
| 10. | Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a |
| 11. | Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007 |
| 12. | Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y |
| 13. | Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149 |
| 14. | Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j |
| 15. | Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q |
| 1. | Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriate, E. Curr. Med. Chem. 2005, 12, 887–916. doi:10.2174/0929867053507315 |
| 2. | Estévez-Braun, A.; González, A. G. Nat. Prod. Rep. 1997, 14, 465–475. doi:10.1039/np9971400465 |
| 3. | O'Kennedy, R.; Thornes, R. D., Eds. Coumarins: Biology, Applications and Mode of Action; John Wiley & Sons: Chichester, U.K., 1997. |
| 4. | Forsyth, C. J.; Ying, L.; Chen, J.; La Clair, J. J. J. Am. Chem. Soc. 2006, 128, 3858–3859. doi:10.1021/ja057087e |
| 5. | Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Bioorg. Med. Chem. Lett. 2008, 18, 423–426. doi:10.1016/j.bmcl.2007.09.100 |
| 6. | Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u |
| 7. | Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111 |
| 8. | Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435 |
| 9. | Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127 |
| 10. | Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a |
| 11. | Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007 |
| 12. | Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y |
| 13. | Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149 |
| 14. | Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j |
| 15. | Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q |
| 33. | Teichert, J. F.; Feringa, B. L. Chem. Commun. 2011, 47, 2679–2681. doi:10.1039/c0cc05160h |
| 34. | Huang, X.; Wen, Y.-H.; Zhou, F.-T.; Chen, C.; Xu, D.-C.; Xie, J.-W. Tetrahedron Lett. 2010, 51, 6637–6640. doi:10.1016/j.tetlet.2010.10.055 |
| 35. | Wang, C.; Tunge, J. A. Org. Lett. 2005, 7, 2137–2139. doi:10.1021/ol050466s |
| 36. | Chen, G.; Tokunaga, N.; Hayashi, T. Org. Lett. 2005, 7, 2285–2288. doi:10.1021/ol0507367 |
| 37. | Dixon, D. J.; Ley, S. V.; Rodríguez, F. Angew. Chem., Int. Ed. 2001, 40, 4763–4765. doi:10.1002/1521-3773(20011217)40:24<4763::AID-ANIE4763>3.0.CO;2-D |
| 62. |
Kuwano, R.; Kondo, Y.; Shirahama, T. Org. Lett. 2005, 7, 2973–2975. doi:10.1021/ol0509787
See for related coupling of benzyl acetates with sulfinates. |
| 26. | Ballerini, E.; Minuti, L.; Piermatti, O.; Pizzo, F. J. Org. Chem. 2009, 74, 4311–4317. doi:10.1021/jo9005365 |
| 27. | Richardson, T. I.; Dodge, J. A.; Durst, G. L.; Pfeifer, L. A.; Shah, J.; Wang, Y.; Durbin, J. D.; Krishnan, V.; Norman, B. H. Bioorg. Med. Chem. Lett. 2007, 17, 4824–4828. doi:10.1016/j.bmcl.2007.06.052 |
| 28. | Girotti, R.; Marrocchi, A.; Minuti, L.; Piermatti, O.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2006, 71, 70–74. doi:10.1021/jo051539o |
| 29. | Haight, A. R.; Bailey, A. E.; Baker, W. S.; Cain, M. H.; Copp, R. R.; DeMattei, J. A.; Ford, K. L.; Henry, R. F.; Hsu, M. C.; Keyes, R. F.; King, S. A.; McLaughlin, M. A.; Melcher, L. M.; Nadler, W. R.; Oliver, P. A.; Parekh, S. I.; Patel, H. H.; Seif, L. S.; Staeger, M. A.; Wayne, G. S.; Wittenberger, S. J.; Zhang, W. Org. Process Res. Dev. 2004, 8, 897–902. doi:10.1021/op049889k |
| 30. | Ohkata, K.; Lee, Y. G.; Utsumi, Y.; Ishimaru, K.; Akiba, K. J. Org. Chem. 1991, 56, 5052–5059. doi:10.1021/jo00017a014 |
| 31. | Yamago, S.; Nakamura, E. J. Am. Chem. Soc. 1989, 111, 7285–7286. doi:10.1021/ja00200a072 |
| 32. | Trost, B. M.; Chan, D. M. T. J. Am. Chem. Soc. 1981, 103, 5972–5974. doi:10.1021/ja00409a083 |
| 45. | Kuwano, R. Synthesis 2009, 2009, 1049–1061. doi:10.1055/s-0028-1088001 |
| 47. | Kuwano, R.; Kondo, Y.; Matsuyama, Y. J. Am. Chem. Soc. 2003, 125, 12104–12105. doi:10.1021/ja037735z |
| 60. | Kuwano, R.; Yokogi, M. Chem. Commun. 2005, 5899–5901. doi:10.1039/B513372F |
| 61. | Kuwano, R.; Yokogi, M. Org. Lett. 2005, 7, 945–947. doi:10.1021/ol050078q |
| 62. |
Kuwano, R.; Kondo, Y.; Shirahama, T. Org. Lett. 2005, 7, 2973–2975. doi:10.1021/ol0509787
See for related coupling of benzyl acetates with sulfinates. |
| 25. | Kim, K.-M.; Park, I.-H. Synthesis 2004, 2004, 2641–2644. doi:10.1055/s-2004-831232 |
| 56. | Chowdhury, S.; Georghiou, P. E. Tetrahedron Lett. 1999, 40, 7599–7603. doi:10.1016/S0040-4039(99)01566-X |
| 57. | Chahen, L.; Doucet, H.; Santelli, M. Synlett 2003, 1668–1672. doi:10.1055/s-2003-40994 |
| 58. | Nobre, S. M.; Monteiro, A. L. Tetrahedron Lett. 2004, 45, 8225–8228. doi:10.1016/j.tetlet.2004.09.020 |
| 59. | Yen, S. K.; Koh, L. L.; Huynh, H. V.; Hor, T. S. A. Eur. J. Inorg. Chem. 2009, 4288–4297. doi:10.1002/ejic.200900447 |
| 60. | Kuwano, R.; Yokogi, M. Chem. Commun. 2005, 5899–5901. doi:10.1039/B513372F |
| 61. | Kuwano, R.; Yokogi, M. Org. Lett. 2005, 7, 945–947. doi:10.1021/ol050078q |
| 16. | Perkin, W. H. J. Chem. Soc. 1868, 21, 53–63. doi:10.1039/JS8682100053 |
| 17. | Woodruff, E. H. Org. Synth. 1955, Coll. Vol. 3, 581. |
| 18. | Kelkar, R. M.; Joshi, U. K.; Paradkar, M. V. Synthesis 1986, 214–216. doi:10.1055/s-1986-31620 |
| 19. | Armstrong, V.; Soto, O.; Valderrama, J. A.; Tapia, R. Synth. Commun. 1988, 18, 717–725. doi:10.1080/00397918808077361 |
| 20. | Darvatkar, N. B.; Deorukhkar, A. R.; Bhilare, S. V.; Raut, D. G.; Salunkhe, M. M. Synth. Commun. 2008, 38, 3508–3513. doi:10.1080/00397910802162967 |
| 21. | Sato, K.; Amakasu, T.; Abe, S. J. Org. Chem. 1964, 29, 2971–2972. doi:10.1021/jo01033a040 |
| 22. | Hepworth, J. D. In Comprehensive Heterocyclic Chemistry; Katritzky, A.; Rees, C. W.; Boulton, A. J.; McKillop, A., Eds.; Pergamon: Oxford, U.K., 1984; Vol. 3, pp 799–810. |
| 23. | Zeitler, K.; Rose, C. A. J. Org. Chem. 2009, 74, 1759–1762. doi:10.1021/jo802285r |
| 24. | Ramachandra, M. S.; Subbaraju, G. V. Synth. Commun. 2006, 36, 3723–3727. doi:10.1080/10916460600946154 |
| 47. | Kuwano, R.; Kondo, Y.; Matsuyama, Y. J. Am. Chem. Soc. 2003, 125, 12104–12105. doi:10.1021/ja037735z |
| 45. | Kuwano, R. Synthesis 2009, 2009, 1049–1061. doi:10.1055/s-0028-1088001 |
| 46. | Weaver, J. D.; Recio, A., III; Grenning, A. J.; Tunge, J. A. Chem. Rev. 2011, 111, 1846–1913. doi:10.1021/cr1002744 |
| 47. | Kuwano, R.; Kondo, Y.; Matsuyama, Y. J. Am. Chem. Soc. 2003, 125, 12104–12105. doi:10.1021/ja037735z |
| 48. | Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2012, 134, 5778–5781. doi:10.1021/ja301461p |
| 49. | Zhu, Y.; Rawal, V. H. J. Am. Chem. Soc. 2011, 134, 111–114. doi:10.1021/ja2095393 |
| 50. | Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2010, 12, 1360–1363. doi:10.1021/ol1002576 |
| 51. | Legros, J.-Y.; Toffano, M.; Fiaud, J.-C. Tetrahedron 1995, 51, 3235–3246. doi:10.1016/0040-4020(95)00061-C |
| 52. | Legros, J.-Y.; Fiaud, J.-C. Tetrahedron Lett. 1992, 33, 2509–2510. doi:10.1016/S0040-4039(00)92227-5 |
| 53. | Torregrosa, R. R. P.; Ariyarathna, Y.; Chattopadhyay, K.; Tunge, J. A. J. Am. Chem. Soc. 2010, 132, 9280–9282. doi:10.1021/ja1035557 |
| 54. | Fields, W. H.; Chruma, J. J. Org. Lett. 2010, 12, 316–319. doi:10.1021/ol902651j |
| 55. | Recio, A., III; Heinzman, J. D.; Tunge, J. A. Chem. Commun. 2012, 48, 142–144. doi:10.1039/c1cc16011g |
| 54. | Fields, W. H.; Chruma, J. J. Org. Lett. 2010, 12, 316–319. doi:10.1021/ol902651j |
| 6. | Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u |
| 7. | Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111 |
| 8. | Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435 |
| 9. | Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127 |
| 10. | Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a |
| 11. | Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007 |
| 12. | Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y |
| 13. | Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149 |
| 14. | Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j |
| 15. | Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q |
| 46. | Weaver, J. D.; Recio, A., III; Grenning, A. J.; Tunge, J. A. Chem. Rev. 2011, 111, 1846–1913. doi:10.1021/cr1002744 |
| 53. | Torregrosa, R. R. P.; Ariyarathna, Y.; Chattopadhyay, K.; Tunge, J. A. J. Am. Chem. Soc. 2010, 132, 9280–9282. doi:10.1021/ja1035557 |
| 54. | Fields, W. H.; Chruma, J. J. Org. Lett. 2010, 12, 316–319. doi:10.1021/ol902651j |
| 55. | Recio, A., III; Heinzman, J. D.; Tunge, J. A. Chem. Commun. 2012, 48, 142–144. doi:10.1039/c1cc16011g |
| 6. | Stefanachi, A.; Favia, A. D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2011, 54, 1613–1625. doi:10.1021/jm101120u |
| 7. | Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. doi:10.1016/j.bmcl.2009.10.111 |
| 8. | Gümüş, A.; Karadeniz, Ş.; Uğraş, H. İ.; Bulut, M.; Çakır, Ü.; Gören, A. C. J. Heterocycl. Chem. 2010, 47, 1127–1133. doi:10.1002/jhet.435 |
| 9. | Pisani, L.; Muncipinto, G.; Miscioscia, T. F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; Passeleu, C.; Carotti, A. J. Med. Chem. 2009, 52, 6685–6706. doi:10.1021/jm9010127 |
| 10. | Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Biochemistry 2009, 48, 10743–10751. doi:10.1021/bi901061a |
| 11. | Kalkhambkar, R. G.; Kulkarni, G. M.; Kamanavalli, C. M.; Premkumar, N.; Asdaq, S. M. B.; Sun, C. M. Eur. J. Med. Chem. 2008, 43, 2178–2188. doi:10.1016/j.ejmech.2007.08.007 |
| 12. | Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D. E.; Mattevi, A. J. Med. Chem. 2007, 50, 5848–5852. doi:10.1021/jm070677y |
| 13. | Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J. J. Med. Chem. 2005, 48, 6400–6408. doi:10.1021/jm0580149 |
| 14. | Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R. W.; Carotti, A. J. Med. Chem. 2004, 47, 6792–6803. doi:10.1021/jm049535j |
| 15. | Onderwater, R. C. A.; Venhorst, J.; Commandeur, J. N. M.; Vermeulen, N. P. E. Chem. Res. Toxicol. 1999, 12, 555–559. doi:10.1021/tx980248q |
| 38. | Wunderlich, S. H.; Rohbogner, C. J.; Unsinn, A.; Knochel, P. Org. Process Res. Dev. 2010, 14, 339–345. doi:10.1021/op9002888 |
| 39. | Martins, S.; Branco, P. S.; de la Torre, M. C.; Sierra, M. A.; Pereira, A. Synlett 2010, 2918–2922. doi:10.1055/s-0030-1259014 |
| 40. | Jeganmohan, M.; Knochel, P. Angew. Chem., Int. Ed. 2010, 49, 8520–8524. doi:10.1002/anie.201003558 |
| 41. | Huang, B.; Liu, J.; Huang, W. J. Chem. Soc., Chem. Commun. 1990, 1781–1782. doi:10.1039/C39900001781 |
| 42. | Jana, R.; Partridge, J. J.; Tunge, J. A. Angew. Chem., Int. Ed. 2011, 50, 5157–5161. doi:10.1002/anie.201100765 |
| 43. | Jana, R.; Trivedi, R.; Tunge, J. A. Org. Lett. 2009, 11, 3434–3436. doi:10.1021/ol901288r |
| 44. | Chattopadhyay, K.; Jana, R.; Day, V. W.; Douglas, J. T.; Tunge, J. A. Org. Lett. 2010, 12, 3042–3045. doi:10.1021/ol101042x |
| 53. | Torregrosa, R. R. P.; Ariyarathna, Y.; Chattopadhyay, K.; Tunge, J. A. J. Am. Chem. Soc. 2010, 132, 9280–9282. doi:10.1021/ja1035557 |
| 55. | Recio, A., III; Heinzman, J. D.; Tunge, J. A. Chem. Commun. 2012, 48, 142–144. doi:10.1039/c1cc16011g |
© 2012 Chattopadhyay et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)