Abstract
Cyclododecane adopts a square-like structure with corner and edge CH2 groups. In this study erythro- and threo-1,2-difluorocyclododecanes were prepared to explore whether the two vicinal C–F bonds, with different relative configurations, preferably locate at corner/edge or edge/edge locations. Conformational analysis comparing the diastereoisomers was explored by using a combination of 19F{1H} NMR spectroscopy, computational studies and, in the case of the threo isomer, X-ray structural analysis. In the lowest energy conformers for both diastereoisomers the vicinal C–F bonds are located corner/edge, rather than edge/edge. These structures avoid placing a C–F bond endo into the ring, and appear to benefit from C–CHF–C angle widening, which relaxes 1,4-H,H transannular interactions.

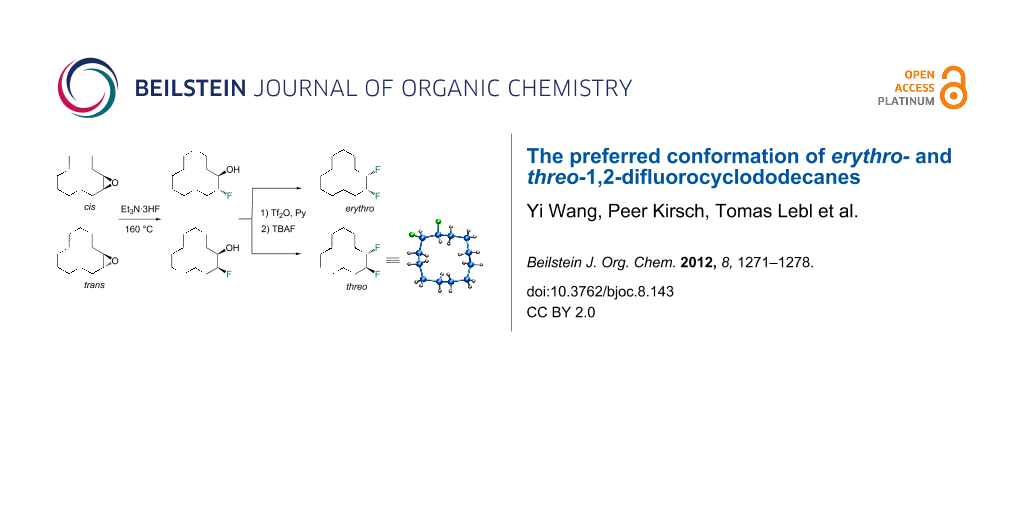
Graphical Abstract
Introduction
The conformation of cyclododecane (1) in the solid state was first reported by Dunitz and Shearer in 1960 [1,2]. They showed that cyclododecane has a square topology, which can be classified as a [3333] type structure [3,4]. Their conclusion was derived from X-ray diffraction data, which could not fully resolve the structure due to a high level of disorder, but the diffraction data was used as the basis of a further computational analysis, and the structure in Figure 1 emerged as their consensus structure [5,6]. The structure is tensioned by transannular interactions in which there are four endo hydrogens, one on each edge pointing into the ring on the top face, and four more endo hydrogens pointing into the ring on the lower face. Each set of hydrogens forms a square, and they are spaced at van der Waals contact distances (~2.1–2.2 Å) to their contacted neighbours, situated at 1,4-positions on adjacent edges of the ring. This structure has two distinct types of methylene group, those located at corners of the ring and those at edges.
![[1860-5397-8-143-1]](/bjoc/content/figures/1860-5397-8-143-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The Dunitz and Shearer structure of cyclododecane (1) [1,2]. There are four endo hydrogens above and four below the plane of the ring, which are tensioned as they are within van der Waals contact distances. Thus the ring adopts a [3333] square-type structure. (Reproduced with permission from [1]; Copyright © 1960 Verlag GmbH & Co KGaA, Weinheim).
Figure 1: The Dunitz and Shearer structure of cyclododecane (1) [1,2]. There are four endo hydrogens above and fou...
Recently we prepared and explored the conformation of cyclododecane ring systems carrying either one or two difluoromethylene (CF2) groups in place of methylenes (CH2) [7]. The study revealed that the CF2 groups always occupied corner positions. This was deduced by a combination of 19F NMR, X-ray structure analysis and theory studies. Two reasons emerged for this. Firstly, if a C–F bond did project into the ring (endo), then the increased size of the fluorine atom relative to hydrogen increases the transannular 1,4-H,F relative to 1,4-H,H strain, by placing a fluorine in the square of hydrogen atoms on the top (or bottom) face of the ring. Secondly, the electronegativity of the fluorines within the CF2 group has the effect of distorting the sp3 geometry and widening the tetrahedral C–CF2–C angle from around 113° to about 116–118° [7-9]. This angle widening has the effect of lengthening/relaxing the 1,4-H,H transannular contacts between the transverse endo hydrogens, thus leading to a lower-energy ring structure. As a consequence of these two effects, the preference for a corner over an edge location is very strong, and thus when the CF2 groups are spaced 1,4 to each other or 1,7 to each other within the ring, they occupy adjacent and opposite corners of the ring, respectively, and form stable ring systems. This is illustrated in the X-ray structures in Figure 2. However when the CF2 groups were placed 1,6 to each other, a deliberate mismatch with respect to corner locations, the ring structure then became a distorted [4332] ring system, which is a structure that is more achievable than placing a CF2 group at an edge position of a [3333] ring system.
![[1860-5397-8-143-2]](/bjoc/content/figures/1860-5397-8-143-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structures of (a) 1,1,4,4- (b) 1,1,7,7- and (c) 1,1,6,6-tetrafluorocyclododecanes (2–4) , illustrating the corner preference of the CF2 groups. Structures 2 and 3 adopt a [3333] conformation, whereas 4 adopts a distorted [4332] conformation. Fluorine atoms are presented in green [7].
Figure 2: Crystal structures of (a) 1,1,4,4- (b) 1,1,7,7- and (c) 1,1,6,6-tetrafluorocyclododecanes (2–4) , i...
In this study we separated the geminal fluorine atom of the CF2 group to generate vicinal fluorines in order to explore the conformational preference of the erythro- and threo- diastereoisomers of 1,2-difluorocyclododecanes 5a and 5b (Figure 3).
![[1860-5397-8-143-3]](/bjoc/content/figures/1860-5397-8-143-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Erythro- and threo-1,2-difluorocyclododecanes (5a and 5b).
Figure 3: Erythro- and threo-1,2-difluorocyclododecanes (5a and 5b).
In the case of erythro- and threo-1,2-difluorocyclododecanes (5a and 5b, respectively) it may be expected that the threo isomer 5b will adopt a conformation whereby the vicinal fluorines occupy a central edge/edge location, such that both C–F bonds project exo to the ring, and do not increase the torsional strain. Also for the threo isomer 5b, the C–F bonds may benefit from hyperconjugative interactions with the anti-periplanar C–H bonds, similar to that found in 1,2-difluoroethane in the well know gauche effect [10,11]. On the other hand, the erythro stereoisomer 5a would have a C–F bond projecting into the ring in an endo manner, if the vicinal fluorines were edge/edge located, and thus it is anticipated that the erythro isomer 5a will adopt a corner/edge location for the C–F bonds rather than an edge/edge location.
In order to test these hypotheses it was necessary to prepare different the diastereoisomers, erythro- and threo-1,2-difluorocyclododecane, and then subject them to conformational analysis by 19F NMR and X-ray structural analyses. A computational study was also carried out to explore the relative energies of the candidate edge/edge and edge/corner conformers.
Results and Discussion
Synthesis
The synthetic route to erythro- (5a) and threo-1,2-difluorocyclododecanes (5b) is shown in Scheme 1. A (1:9) mixture of cis- and trans-epoxides was treated with triethylamine trihydrofluoride [12]. This afforded diastereoisomeric fluorohydrins 7a and 7b, which could be readily separated by chromatography. Each fluorohydrin was then treated with triflic anhydride [13,14], to generate the corresponding triflates 8a and 8b. Treatment of 8a and 8b with tetrabutylammonium fluoride (TBAF) in THF was stereospecific and independently generated the erythro- or threo-1,2-difluorocyclododecanes 5a and 5b. These compounds were white solids. In the case of the threo isomer only, a suitable crystal was grown such that an X-ray structure could be solved. The resultant structure, which confirmed the threo stereochemistry, is shown in Figure 4. Notably one of the C–F bonds occupies a corner location, inconsistent with our preconceived expectation of a edge/edge conformation. This corner/edge conformation appears to be favoured in the solid state over the edge/edge conformation. Also the C–CHF–C angle of 117.0° indicates a small rehybridisation tendency towards a wider angle, observed more dramatically with the CF2 group [7-9], and with concomitant release of angle strain.
![[1860-5397-8-143-i1]](/bjoc/content/inline/1860-5397-8-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to erythro- (5a) and threo-1,2-difluorocyclododecane (5b).
Scheme 1: Synthetic routes to erythro- (5a) and threo-1,2-difluorocyclododecane (5b).
![[1860-5397-8-143-4]](/bjoc/content/figures/1860-5397-8-143-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of threo-1,2-difluorocyclododecane (5b) showing corner angles and representative transannular contact distances.
Figure 4: X-ray crystal structure of threo-1,2-difluorocyclododecane (5b) showing corner angles and represent...
Variable-temperature (VT) 19F NMR
Low-temperature (CD2Cl2, 180 K) 19F{1H} NMR experiments were carried out on both the erythro and threo isomers. The spectra are shown in Figure 5. In each case at room temperature there is a single fluorine resonance; however, on lowering of the temperature the fluorine signal resolves into an AB-system, indicating nonequivalent fluorine environments and, thus, corner/edge locations of the C–F bonds in each case. Vicinal edge/edge conformations would result in the magnetic equivalence of the fluorine atoms, but this is not observed. Both isomers 5a and 5b display a single resonance at room temperature (25 °C), indicating rapid ring interconversion on the NMR timescale. Rate constants for the ring interconversions were determined by complete lineshape analysis of the 19F NMR spectra recorded across the temperature range 180–295 K.
![[1860-5397-8-143-5]](/bjoc/content/figures/1860-5397-8-143-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Variable-temperature 19F{1H} NMR of erythro- (5a) and threo-1,2-difluorocyclododecane (5b).
Figure 5: Variable-temperature 19F{1H} NMR of erythro- (5a) and threo-1,2-difluorocyclododecane (5b).
Fitting the experimental data to the Eyring equation [15] allowed determination of the activation parameters (see Table 1 and Supporting Information File 1).
The overall free energy change (ΔG#) is similar in each case and both the erythro 5a and threo 5b stereoisomers have conformational energy barriers ~2–3 kcal·mol−1 higher than cyclododecane itself (7.3 kcal·mol−1) indicating that fluorine introduces some conformational stability. The enthalpy difference (ΔH#) is significant between the isomers. The theory calculations described below suggest that the erythro isomer 5a is more stable than the threo isomer 5b in the ground state, thus this is most probably the major contributor to the enthalpy difference. The opposite sign in entropy (ΔS#) for each isomer makes a relatively small contribution to the overall free energy; however, the positive value for the erythro isomer is perhaps unexpected for progression towards a transition state. This may arise as a result of desolvation for this isomer.
Computational study
In order to explore conformer energies further, a theoretical study MP2/6-311+G(2d,p)//B3LYP/6-311+G(2d,p)+ZPE) [16] was carried out to assess relative ground-state energies of candidate conformers. The structures and relative energies for the erythro 5a and threo 5b isomers are shown in Figure 6. These data indicate that the corner/edge conformers are more stable than the alternative edge/edge conformers for each stereoisomer. This is consistent with the conclusions from the experimental VT 19F NMR study.
![[1860-5397-8-143-6]](/bjoc/content/figures/1860-5397-8-143-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Calculated relative energies of the conformations of the erythro (5a) and threo (5b) stereoisomers of 1,2-difluorocyclododecane.
Figure 6: Calculated relative energies of the conformations of the erythro (5a) and threo (5b) stereoisomers ...
For the erythro stereoisomer 5a, three conformers I–III were considered. Conformers I and II each have a fluorine pointing into the ring (endo), and thus there is an increase in transannular ring strain, raising the energy of these conformers by 2.81 and 3.72 kcal·mol−1 respectively above conformer III, the lowest in energy. For the threo stereoisomer 5b, four conformers, IV–VII were considered. Conformers IV and V, which have two and one endo fluorine, respectively, are highest in energy. In particular, conformer IV with two endo fluorines has a ground-state energy of 6.65 kcal·mol−1, the highest of all of those examined, illustrating the additive and negative impact of placing fluorines into endo orientations. Conformers VI and VII are lower in energy. It was anticipated at the outset that conformer VI may be the most favoured for the threo isomer; however, this does not appear to be the case, although the energy difference between the lowest-energy corner/edge conformer VII and edge/edge conformer VI is relatively small at VI − VII = 0.7 kcal·mol−1. This theoretical observation is supported by the VT 19F{1H} NMR study, which indicates nonequivalent fluorines consistent with a corner location. Also the structure of conformer VII is almost identical to that obtained experimentally by X-ray structure analysis (Figure 4).
It is not immediately obvious why threo 5b conformer VII is favoured (lower in energy) over conformer VI, although the energy difference is small (~0.7 kcal·mol−1). It is noteworthy that the fluorine atoms are a little closer in intramolecular distance in VII (F···F 2.68Å) compared to VI (F···F 2.75Å), and thus electrostatic repulsion does not appear to be the discriminating factor. One origin for this preference, which emerges from the theoretical study, may be the widening of the C−CHF−C angle (115.62°) in VII. The introduction of fluorine alters the hybridisation at carbon [5], and this should relieve angular strain in these tensioned ring systems relative to a strained C−CH2−C angle of ~115° (see Table 2).
Table 2: The corner C–C–C angles for conformers VI and VII of the threo isomer 5b. The C–CHF–C angle (115.62°) is the largest; however some C–CH2–C angles are clearly strained at ~115.0°.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-143-i2.png?max-width=637&scale=1.0)
|
||||
| threo 5b | top left corner [°] | top right corner [°] | bottom left corner [°] | bottom right corner [°] |
|---|---|---|---|---|
| VI | 113.42 (CH2) | 113.41 (CH2) | 115.16 (CH2) | 115.21 (CH2) |
| VII | 115.62 (CHF) | 113.91 (CH2) | 115.09 (CH2) | 114.32 (CH2) |
Conclusion
The erythro (5a) and threo (5b) isomers of 1,2-difluorocyclododecane were synthesised, and their preferred conformers were explored experimentally by 19F NMR, and X-ray structure analysis. A computational study was also carried out to establish favoured conformations and their relative ground-state energies. A particular focus of the study examined whether the vicinal C–F bonds prefer to adopt corner/edge or edge/edge locations of the [3333] ring system. For each diastereoisomer it emerged that one of the C–F bonds adopts a corner location. The second orientates exo to the ring. When the C–F bond projects endo into the ring, the energy of the system is raised by between 2.0–3.0 kcal·mol−1 and is disfavoured. In the case of the threo isomer 5b, the outcome was less easy to predict, as an edge/edge structure can be achieved with both C–F bonds exo to the ring, and with each C–F bond benefiting from antiperiplanar C–H/C–F hyperconjugative interactions. However it would appear that the corner/edge structure is still favoured for the threo isomer 5b, as the C–C–C bond angles of ~115°, which occur in these tensioned ring systems, are inherently less strained, due to rehybridisation/angle widening, if they carry a central fluorine atom.
Experimental
Preparation of 7a and 7b: Commercially available 1,2-epoxycyclododecane (6) (5 mmol, 0.91 g, 9:1 trans/cis) and Et3N·3HF (4.0 g, 25 mmol) was added to a Teflon-coated reactor and stirred at 160 °C for 24 h. After cooling down, the reaction mixture was quenched with sat. NaHCO3 solution (50 mL) and extracted into diethyl ether (3 × 20 mL). The organic layers were combined and dried (MgSO4), and then concentrated under vacuum. Purification over silica gel, eluting with hexane and diethyl ether (90:10), yielded trans-2-fluorocyclododecanol (7a) (91 mg, 9%) and cis-2-fluorocyclododecanol (7b) (420 mg, 41%) as a white crystalline solid.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-143-i3.svg?max-width=637&scale=1.18182)
7a: mp 64–65 °C; 1H NMR (400 MHz, CDCl3) δH 4.56 (ddt, J = 49.4, 8.7, 4.2 Hz, CHF), 3.94–3.86 (m, 1H, CHOH), 2.27 (t, J = 3.5 Hz, 1H, CHOH) and 1.92–0.84 (m, 20H, 10 × CH2); 13C NMR (100 MHz, CDCl3) δC 95.3 (d, J = 166 Hz, CHF), 71.4 (d, J = 18 Hz, CHOH), 28.7 (d, J = 5.5 Hz, CH2), 27.8 (d, J = 21 Hz, CH2), 24.0 (d, J = 1.6 Hz, 2 × CH2), 23.7 (d, J = 2.8 Hz, 2 × CH2), 22.6 (d, J = 2.9 Hz, 2 × CH2), 20.6 (CH2), 20.4 (d, J = 2.8 Hz, CH2); {1H}19F NMR (376 MHz, CDCl3) δF −194.0 (CHF); LRMS–ESI (m/z): [M + Na]+ calcd for C12H23OFNa, 225.16; found, 225.06.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-143-i4.svg?max-width=637&scale=1.18182)
7b: mp 84–86 °C; 1H NMR (400 MHz, CDCl3) δH 4.70 (dtd, J = 47.3, 6.0, 1.7 Hz, 1H, CHF), 3.99–3.88 (m, 1H, CHOH), 1.81 (d, J = 5.4 Hz, 1H, CHOH) and 1.83–1.33 (m, 20H, 10 × CH2); 13C NMR (75 MHz, CDCl3) δC 95.4 (d, J = 168 Hz, CHF), 71.6 (d, J = 20 Hz, CHOH), 28.3 (d, J = 7 Hz, CH2), 25.2 (d, J = 21 Hz, CH2), 24.6 (CH2), 24.3 (CH2), 23.8 (CH2), 23.6 (CH2), 21.7 (2 × CH2), 21.4 (d, J = 5 Hz, CH2), 21.3 (CH2); {1H}19F NMR (376 MHz, CDCl3) δF −191.1 (CHF); LRMS–ESI (m/z): [M + Na]+ calcd for C12H23OFNa, 225.16; found, 225.07.
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-143-i5.svg?max-width=637&scale=1.18182)
Erythro isomer 5a: To a solution of 7a (0.11 g, 0.5 mmol) in DCM (5 mL) at 0 °C was added pyridine (80 µL, 1 mmol, 2 equiv) and Tf2O (130 µL, 0.7 mmol, 1.4 equiv). The resulting mixture was stirred for 1 h and the solvent was removed under vacuum. The residue containing the trans-fluorotriflate 8a was dissolved in THF (3 mL), and TBAF solution (1 mL, 1 M in THF, 1 mmol) was added dropwise. The reaction mixture was stirred at rt for 48 h and monitored by 19F NMR. Purification over silica gel, eluting with 1% diethyl ether in cyclohexane yielded erythro-difluorocyclododecane 5a (21 mg, 21%) as a white solid: mp 57 °C; 1H NMR (400 MHz, CDCl3) δH 4.81–4.64 (tdd, J = 48.3, 24.1, 6.3 Hz, 2H, 2 × CHF), 1.80–1.73 (m, 4H, 2 × CH2), 1.44–1.33 (m, 16H, 8 × CH2); 13C NMR (75 MHz, CDCl3) δC 92.5 (dd, J = 174, 20 Hz, 2 × CHF), 26.9 (2 × CH2), 25.6 (dd, J = 21, 6 Hz, CH2), 24.0 (4 × CH2), 21.6 (2 × CH2), 21.0 (CH2), 20.9 (CH2); {1H}19F NMR (376 MHz, CDCl3) δF −191.0 (CF); HRMS–ESI (m/z): [M + Na]+ exact mass calcd for C12H22F2Na, 227.1587; found, 227.1591.
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-143-i6.svg?max-width=637&scale=1.18182)
Threo isomer 5b: Similar to the erythro compound, 5b was obtained from cis-7b in 25% as a white solid: mp 61 °C; 1H NMR (400 MHz, CDCl3) δH 4.81–4.64 (m, 2H, 2 × CHF), 2.10–1.23 (m, 20H, 10 × CH2); 13C NMR (100 MHz, CD2Cl2) δC 92.4 (dd, J = 176, 19 Hz, 2 × CHF), 29.7 (CH2), 27.8 (dd, J = 15, 12 Hz, 2 × CH2), 24.0 (2 × CH2), 23.7 (2 × CH2), 22.5 (2 × CH2), 20.4 (CH2); {1H}19F NMR (376 MHz, CDCl3) δF −193.6 (CF); HRMS–ESI (m/z): [M + Na]+ exact mass calcd for C12H22F2Na, 227.1587; found, 227.1595.
Supporting Information
The Supporting Information contains NMR spectra and results of the differential scanning calorimetry, variable-temperature NMR, and computational studies.
| Supporting Information File 1: Additional data. | ||
| Format: PDF | Size: 2.2 MB | Download |
References
-
Dunitz, J. D.; Shearer, H. M. M. Helv. Chim. Acta 1960, 43, 18–35. doi:10.1002/hlca.19600430104
Return to citation in text: [1] [2] [3] -
Bürgi, H.-B.; Dunitz, J. D. Helv. Chim. Acta 1993, 76, 1115–1166. doi:10.1002/hlca.19930760303
Return to citation in text: [1] [2] -
Dale, J. Acta Chem. Scand. 1973, 27, 1115–1129. doi:10.3891/acta.chem.scand.27-1115
Return to citation in text: [1] -
Eliel, E. L.; Wilen, S. H. Stereochemistry of Organic Compounds; Wiley-Interscience: New York, 1994.
Return to citation in text: [1] -
Anet, F. A. L.; Rawdah, T. N. J. Am. Chem. Soc. 1978, 100, 7166–7171. doi:10.1021/ja00491a007
Return to citation in text: [1] [2] -
Atavin, E. G.; Mastryukov, V. S.; Allinger, N. L.; Almenningen, A.; Seip, R. J. Mol. Struct. 1989, 212, 87–95. doi:10.1016/0022-2860(89)85069-0
Return to citation in text: [1] -
Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060
Return to citation in text: [1] [2] [3] [4] -
Dasaradhi, L.; O'Hagan, D.; Petty, M. C.; Pearson, C. J. Chem. Soc., Perkin Trans. 2 1995, 221–225. doi:10.1039/p29950000221
Return to citation in text: [1] [2] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] [2] -
Goodman, L.; Gu, H.; Pophristic, V. J. Phys. Chem. A 2005, 109, 1223–1229. doi:10.1021/jp046290d
Return to citation in text: [1] -
Souza, F. R.; Freitas, M. P.; Rittner, R. J. Mol. Struct. 2008, 863, 137–140. doi:10.1016/j.theochem.2008.06.003
Return to citation in text: [1] -
Michel, D.; Schlosser, M. Tetrahedron 2000, 56, 4253–4260. doi:10.1016/S0040-4020(00)00351-3
Return to citation in text: [1] -
Nicoletti, M.; O'Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2005, 127, 482–483. doi:10.1021/ja045299q
Return to citation in text: [1] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O'Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/c1cc13016a
Return to citation in text: [1] -
Eyring, H. J. Chem. Phys. 1935, 3, 107–115. doi:10.1063/1.1749604
Return to citation in text: [1] -
Gaussian 03, Revision D.01: M. J. Frisch et al. [see Supporting Information], Gaussian, Inc., Wallingford CT, 2004. The minimum geometries were optimised at the B3LYP/6-311+G(2d,p) level of theory, and were verified to have only positive eigenfrequencies. The energies of the conformers were calculated at the MP2/6-311+G(2d,p) level of theory, by using both B3LYP/6-311+G(2d,p) and M06-2X/6-311+G(2d,p) geometries and zero-point energies. There are slight energy differences but no qualitative changes in the relative energies.
Return to citation in text: [1]
| 1. | Dunitz, J. D.; Shearer, H. M. M. Helv. Chim. Acta 1960, 43, 18–35. doi:10.1002/hlca.19600430104 |
| 2. | Bürgi, H.-B.; Dunitz, J. D. Helv. Chim. Acta 1993, 76, 1115–1166. doi:10.1002/hlca.19930760303 |
| 1. | Dunitz, J. D.; Shearer, H. M. M. Helv. Chim. Acta 1960, 43, 18–35. doi:10.1002/hlca.19600430104 |
| 5. | Anet, F. A. L.; Rawdah, T. N. J. Am. Chem. Soc. 1978, 100, 7166–7171. doi:10.1021/ja00491a007 |
| 1. | Dunitz, J. D.; Shearer, H. M. M. Helv. Chim. Acta 1960, 43, 18–35. doi:10.1002/hlca.19600430104 |
| 2. | Bürgi, H.-B.; Dunitz, J. D. Helv. Chim. Acta 1993, 76, 1115–1166. doi:10.1002/hlca.19930760303 |
| 5. | Anet, F. A. L.; Rawdah, T. N. J. Am. Chem. Soc. 1978, 100, 7166–7171. doi:10.1021/ja00491a007 |
| 6. | Atavin, E. G.; Mastryukov, V. S.; Allinger, N. L.; Almenningen, A.; Seip, R. J. Mol. Struct. 1989, 212, 87–95. doi:10.1016/0022-2860(89)85069-0 |
| 3. | Dale, J. Acta Chem. Scand. 1973, 27, 1115–1129. doi:10.3891/acta.chem.scand.27-1115 |
| 4. | Eliel, E. L.; Wilen, S. H. Stereochemistry of Organic Compounds; Wiley-Interscience: New York, 1994. |
| 16. | Gaussian 03, Revision D.01: M. J. Frisch et al. [see Supporting Information], Gaussian, Inc., Wallingford CT, 2004. The minimum geometries were optimised at the B3LYP/6-311+G(2d,p) level of theory, and were verified to have only positive eigenfrequencies. The energies of the conformers were calculated at the MP2/6-311+G(2d,p) level of theory, by using both B3LYP/6-311+G(2d,p) and M06-2X/6-311+G(2d,p) geometries and zero-point energies. There are slight energy differences but no qualitative changes in the relative energies. |
| 10. | Goodman, L.; Gu, H.; Pophristic, V. J. Phys. Chem. A 2005, 109, 1223–1229. doi:10.1021/jp046290d |
| 11. | Souza, F. R.; Freitas, M. P.; Rittner, R. J. Mol. Struct. 2008, 863, 137–140. doi:10.1016/j.theochem.2008.06.003 |
| 13. | Nicoletti, M.; O'Hagan, D.; Slawin, A. M. Z. J. Am. Chem. Soc. 2005, 127, 482–483. doi:10.1021/ja045299q |
| 14. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O'Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/c1cc13016a |
| 7. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 7. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 8. | Dasaradhi, L.; O'Hagan, D.; Petty, M. C.; Pearson, C. J. Chem. Soc., Perkin Trans. 2 1995, 221–225. doi:10.1039/p29950000221 |
| 9. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 7. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 8. | Dasaradhi, L.; O'Hagan, D.; Petty, M. C.; Pearson, C. J. Chem. Soc., Perkin Trans. 2 1995, 221–225. doi:10.1039/p29950000221 |
| 9. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 7. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 12. | Michel, D.; Schlosser, M. Tetrahedron 2000, 56, 4253–4260. doi:10.1016/S0040-4020(00)00351-3 |
© 2012 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)