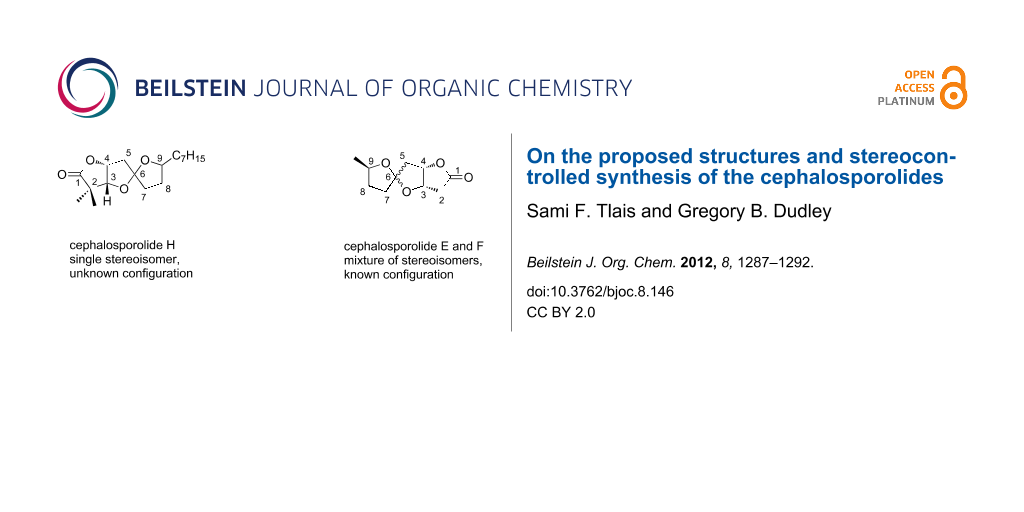
Abstract
The synthesis of four candidate stereoisomers of cephalosporolide H is described, made possible by a zinc-chelation strategy for controlling the stereochemistry of oxygenated 5,5-spiroketals. The same strategy likewise enables the first stereocontrolled synthesis of cephalosporolide E, which is typically isolated and prepared admixed with its spiroketal epimer, cephalosporolide F.

Graphical Abstract
Introduction
The spiroketal moiety is common in natural products of marine, plant, insect, and bacterial origins [1-11]. The rigidity of the spiroketal provides defined orientation of pendant functional groups, and there is a strong correlation between bioactivity and spiroketal stereochemistry in many natural spiroketals. For example, cephalostatin and ritterazine feature thermodynamically disfavored spiroketals that are more cytotoxic than their stereoisomers [12]. Other prominent cytotoxic spiroketals include spongistatin [7] and norhalichondrin [13,14].
Spiroketal-containing pheromones are especially prominent in insect communication, with the spiroketal stereochemistry often relaying important information [15]. For example, the R-enantiomer of the olive-fly sex pheromone is attractive to males, while the S-enantiomer attracts females (Figure 1). Chalcogran, a sex pheromone secreted by the male bark beetle, is isolated from natural sources as a mixture of diastereomers. However, Byers et al showed that the spiroketal (2R,5R)-isomer induces the strongest responses from both females and males [16]. In such cases it is interesting to conjecture that the natural pheromone signal may include a “time stamp”: release of a stereodefined, nonthermodynamic and labile spiroketal enables the detecting insect to gauge the age of the message based on the integrity of the spiroketal center. To duplicate such communication (e.g., for insect population control) requires the ability to prepare specific spiroketal stereoisomers. Anomeric effects typically guide the stereochemical preferences in 6,6 and 5,6-spiroketals [7,17-20], whereas 5,5-spiroketal stereochemistry is more difficult to predict [21] and control [22-25].
![[1860-5397-8-146-1]](/bjoc/content/figures/1860-5397-8-146-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
This report focuses on 5,5-spiroketal lactones of the cephalosporolide and related families (Figure 2) [26,27]. Cephalosporolides E and F are co-isolated as a mixture, and previous syntheses likewise produce these compounds as a mixture in the absence of methods to control the spiro-center [28-30]. However, it is quite possible that the microorganism produces one or the other of these isomers selectively, and that this material scrambles over time to a thermodynamic mixture. It is this mixture that is ultimately extracted, and if this conjecture is true, then the natural extract (mixture) would misrepresent the compound as used in the biological system. Cephalosporolide H, I, and the penisporolides, on the other hand, were isolated as single isomers [31]. The structures of these related spiroketals were tentatively proposed based on NOESY experiments, and by analogy to each other and to the confirmed structures of cephalosporolide E and F.
![[1860-5397-8-146-2]](/bjoc/content/figures/1860-5397-8-146-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reported structures of the cephalosporolides and penisporolides.
Figure 2: Reported structures of the cephalosporolides and penisporolides.
Results and Discussion
We recently reported the stereocontrolled synthesis of cephalosporolide H (reported structure) and its spiroketal isomer [32]. Our strategy featured the use of zinc salts to control the spiro-center using either steric biases or chelation, depending on pendant functionality (Scheme 1). Unfortunately, neither of the synthetic isomers provided data in complete agreement with data reported for the natural material.
![[1860-5397-8-146-i1]](/bjoc/content/inline/1860-5397-8-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Stereocontrol of oxygenated 5,5-spiroketals.
Scheme 1: Stereocontrol of oxygenated 5,5-spiroketals.
Here, we report the synthesis of all four possible diastereomers with respect to C9 and the spiroketal center (C6). Data from this study allows us to comment on the proposed structure for cephalosporolide H and, by analogy, cephalosporolide I and the penisporolides. Expanding on the zinc chelation method, we investigated a new approach to the synthesis of cephalosporolide E, which resulted in the first stereocontrolled assembly of this spiroketal core.
Our synthesis of the reported structure of cephalosporolide H (1) and its spiroketal isomer (1b) are outlined in Scheme 2. The synthesis starts from D-pantolactone (4), which was converted into terminal alkyne 5 [32]. Alkyne 5 was coupled with (R)-1,2-epoxynonane to obtain internal alkyne 6, which was submitted to gold-catalyzed cycloisomerization [33] to afford spiroketals 7a and 7b (the silyl ether is concomitantly hydrolyzed) as a 1:1 mixture of isomers. Exposure of this mixture to zinc chloride promoted isomerization to provide 7a in >20:1 dr. TEMPO oxidation then completed the synthesis of 1, the reported structure of cephalosporolide H. The opposite spiroketal isomer 1a was prepared from 6 by palladium-catalyzed cycloisomerization (steric control), desilylation and TEMPO oxidation. In neither case did the characterization data match that of the natural material, and it is worth noting that NOE and NOESY experiments conducted on both isomers were inconclusive in attempts to differentiate them: similar NOESY cross-peaks were observed from both isomers.
![[1860-5397-8-146-i2]](/bjoc/content/inline/1860-5397-8-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the reported cephalosporolide H and its spiro isomer.
Scheme 2: Synthesis of the reported cephalosporolide H and its spiro isomer.
We prepared the two remaining core diastereomers by similar routes (Scheme 3), starting by coupling terminal alkyne 5 with (S)-1,2 epoxynonane. Gold-catalyzed cycloisomerization (with desilylation) provided spiroketal diols 10a and 10b in a 32:68 ratio and in 89% total yield. Major spiroketal 10b could be converted to 10a in 15:1 dr by zinc-catalyzed isomerization. Both isomers (10a and 10b) were independently subjected to TEMPO oxidation to afford spiroketal lactones 1b and 1c. Spectroscopic data of 1b and 1c were similar to 1a and 1, respectively.
![[1860-5397-8-146-i3]](/bjoc/content/inline/1860-5397-8-146-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the reported C9-epi-cephalosporolide H and its spiro isomer.
Scheme 3: Synthesis of the reported C9-epi-cephalosporolide H and its spiro isomer.
Attempts to secure an authentic sample and/or copies of original NMR spectra for the natural material were unsuccessful, but two candidates emerged as good fits to the reported data (Figure 3). Spectroscopic data for 1a and 1b were both nearly consistent with the data reported for cephalosporolide H, whereas data for 1 and 1c did not match (see Supporting Information File 1, Table S1 for a full comparison). In the absence of an authentic sample, a definitive assignment will not be possible, although we note that the relative stereochemistry of 1b corresponds to that of cephalosporolide F, the structure of which has been confirmed.
![[1860-5397-8-146-3]](/bjoc/content/figures/1860-5397-8-146-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Reported and synthesized cephalosporolide H isomers.
Figure 3: Reported and synthesized cephalosporolide H isomers.
A central feature of this study is our ability to prepare the four diastereomers selectively. Specifically, we found that chelation of zinc salts between the spiroketal oxygen and appropriately positioned hydroxyls overrides normal steric biases to guide the formation of the spiroketal. Cephalosporolide E was targeted for validation of this approach. There are three main differences between cephalosporolides E and H (Figure 2): (1) Cephalosporolide E was isolated (and has been prepared [28]) admixed with cephalosporolide F, whereas cephalosporolide H was isolated as single isomer. (2) Cephalosporolide E (and F) has a C2 methylene; C2 of cephalosporolide H is quaternary. (3) Cephalosporolide E (and F) has a methyl group at C9, as opposed to the n-heptyl chain in cephalosporolide H.
Synthesis of cephalosporolide E started with the known alcohol 12, which was prepared from the commercially available diester 11 (Scheme 4) [34]. PMB protection of alcohol 12 followed by Sharpless dihydroxylation afforded diol 14 [35,36]. DDQ oxidation of PMB ether produced 1,3-dioxane 15 [37]. Protecting group manipulation led to the formation of primary alcohol 17 [38], which was converted into homopropargyl silyl ether 19 over two steps, i.e., DMP oxidation and subsequent Ohira–Bestmann ethynylation [39].
![[1860-5397-8-146-i4]](/bjoc/content/inline/1860-5397-8-146-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of homopropargyl silyl ether.
Scheme 4: Synthesis of homopropargyl silyl ether.
Coupling of the propargyl silyl ether 19 with the (R)-propylene oxide produced the internal alkyne 20 (Scheme 5). Gold(I) chloride in MeOH induced the spiroketalization of alkyne 20 with concomitant cleavage of the PMP acetal and partial cleavage of the TBS ether. After completion of the desilylation with TBAF, a mixture of 5,5-spiroketals 21a and 21b was obtained in 71% overall yield from 20. The mixture of diols 21a and 21b converged to 21a (epimer 21b no longer observable by 1H NMR) upon treatment with zinc chloride. TEMPO oxidation of diol 21a led to the formation of cephalosporolide E (2, admixed with a minor diastereomer tracing back to the Sharpless dihydroxylation reaction). Spectroscopic data for our synthetic material was in full agreement with the reported data for cephalosporolide E [28-30].
![[1860-5397-8-146-i5]](/bjoc/content/inline/1860-5397-8-146-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of cephalosporolide E.
Scheme 5: Synthesis of cephalosporolide E.
Conclusion
We have completed the stereocontrolled synthesis of the reported structure of cephalosporolide H and three diastereoisomers, leading us to suggest a potential structure for natural cephalosporolide H (i.e., 1b, or perhaps 1a). Chelation by using zinc chloride plays a key role in accessing the otherwise contra-thermodynamic spiroketal stereoisomers. This strategy was expanded to enable synthetic production of cephalosporolide E for the first time in a stereocontrolled manner.
Supporting Information
| Supporting Information File 1: General experimental procedures, experimental and characterization data for new compounds, copies of NMR spectra, and comparison of characterization data reported for cephalosporolide H and obtained for the four synthetic diastereomers reported here. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Kluge, A. F. Heterocycles 1986, 24, 1699–1740. doi:10.3987/R-1986-06-1699
Return to citation in text: [1] -
Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4
Return to citation in text: [1] -
Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015
Return to citation in text: [1] -
Jacobs, M. F.; Kitching, W. B. Curr. Org. Chem. 1998, 2, 395–436.
Return to citation in text: [1] -
Francke, W.; Kitching, W. Curr. Org. Chem. 2001, 5, 233–251. doi:10.2174/1385272013375652
Return to citation in text: [1] -
Mead, K. T.; Brewer, B. N. Curr. Org. Chem. 2003, 7, 227–256. doi:10.2174/1385272033372969
Return to citation in text: [1] -
Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n
Return to citation in text: [1] [2] [3] -
Raju, B. R.; Saikia, A. K. Molecules 2008, 13, 1942–2038. doi:10.3390/molecules13081942
Return to citation in text: [1] -
Favre, S.; Vogel, P.; Gerber-Lemaire, S. Molecules 2008, 13, 2570–2600. doi:10.3390/molecules13102570
Return to citation in text: [1] -
Sperry, J.; Wilson, Z. E.; Rathwell, D. C. K.; Brimble, M. A. Nat. Prod. Rep. 2010, 27, 1117–1137. doi:10.1039/b911514p
Return to citation in text: [1] -
Ballini, R.; Petrini, M.; Rosini, G. Molecules 2008, 13, 319–330. doi:10.3390/molecules13020319
Return to citation in text: [1] -
LaCour, T. G.; Guo, C.; Bhandaru, S.; Boyd, M. R.; Fuchs, P. L. J. Am. Chem. Soc. 1998, 120, 692–707. doi:10.1021/ja972160p
Return to citation in text: [1] -
Jackson, K. L.; Henderson, J. A.; Motoyoshi, H.; Phillips, A. J. Angew. Chem., Int. Ed. 2009, 48, 2346–2350. doi:10.1002/anie.200806111
Return to citation in text: [1] -
Yamamoto, A.; Ueda, A.; Brémond, P.; Tiseni, P. S.; Kishi, Y. J. Am. Chem. Soc. 2012, 134, 893–896. doi:10.1021/ja2108307
Return to citation in text: [1] -
Mori, K. Bioorg. Med. Chem. 2007, 15, 7505–7523. doi:10.1016/j.bmc.2007.08.040
Return to citation in text: [1] -
Byers, J. A.; Högberg, H.-E.; Unelius, C. R.; Birgersson, G.; Löfqvist, J. J. Chem. Ecol. 1989, 15, 685–695. doi:10.1007/BF01014711
Return to citation in text: [1] -
Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry; Pergamon Press: Oxford, 1983.
Return to citation in text: [1] -
Juaristi, E.; Cuevas, G. The Anomeric Effect; CRC Press: Boca Raton, FL, 1994.
Return to citation in text: [1] -
Lau, C. K.; Crumpler, S.; Macfarlane, K.; Lee, F.; Berthelette, C. Synlett 2004, 2281–2286. doi:10.1055/s-2004-831334
Return to citation in text: [1] -
Evans, D. A.; Coleman, P. J.; Dias, L. C. Angew. Chem., Int. Ed. Engl. 1997, 36, 2738–2741. doi:10.1002/anie.199727381
Return to citation in text: [1] -
Legon, A. C. Chem. Rev. 1980, 80, 231–262. doi:10.1021/cr60325a002
Return to citation in text: [1] -
Fortner, K. C.; Kato, D.; Tanaka, Y.; Shair, M. D. J. Am. Chem. Soc. 2010, 132, 275–280. doi:10.1021/ja906996c
Return to citation in text: [1] -
Phillips, S. T.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 6589–6598. doi:10.1021/ja0705487
Return to citation in text: [1] -
Ravindar, K.; Reddy, M. S.; Lindqvist, L.; Pelletier, J.; Deslongchamps, P. Org. Lett. 2010, 12, 4420–4423. doi:10.1021/ol1019663
Return to citation in text: [1] -
Solladié, G.; Huser, N.; Fischer, J.; Decian, A. J. Org. Chem. 1995, 60, 4988–4990. doi:10.1021/jo00121a015
Return to citation in text: [1] -
Li, X.; Yao, Y.; Zheng, Y.; Sattler, I.; Lin, W. Arch. Pharmacal Res. 2007, 30, 812–815. doi:10.1007/BF02978829
Return to citation in text: [1] -
Ackland, M. J.; Hanson, J. R.; Hitchcock, P. B.; Ratcliffe, A. H. J. Chem. Soc., Perkin Trans. 1 1985, 843–847. doi:10.1039/P19850000843
Return to citation in text: [1] -
Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z
Return to citation in text: [1] [2] [3] -
Fernandes, R. A.; Ingle, A. B. Synlett 2010, 158–160. doi:10.1055/s-0029-1218538
Return to citation in text: [1] [2] -
Brimble, M. A.; Finch, O. C.; Heapy, A. M.; Fraser, J. D.; Furkert, D. P.; O’Connor, P. D. Tetrahedron 2011, 67, 995–1001. doi:10.1016/j.tet.2010.11.107
Return to citation in text: [1] [2] -
Li, X.; Sattler, I.; Lin, W. J. Antibiot. 2007, 60, 191–195. doi:10.1038/ja.2007.21
Return to citation in text: [1] -
Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z
Return to citation in text: [1] [2] -
Tlais, S. F.; Dudley, G. B. Beilstein J. Org. Chem. 2011, 7, 570–577. doi:10.3762/bjoc.7.66
Return to citation in text: [1] -
Hoppen, S.; Bäurle, S.; Koert, U. Chem.–Eur. J. 2000, 6, 2382–2396. doi:10.1002/1521-3765(20000703)6:13<2382::AID-CHEM2382>3.0.CO;2-C
Return to citation in text: [1] -
Brandl, T.; Hoffmann, R. W. Eur. J. Org. Chem. 2002, 2613–2623. doi:10.1002/1099-0690(200208)2002:15<2613::AID-EJOC2613>3.0.CO;2-Y
Return to citation in text: [1] -
Corey, E. J.; Guzman-Perez, A.; Noe, M. C. Tetrahedron Lett. 1995, 36, 3481–3484. doi:10.1016/0040-4039(95)00570-3
Return to citation in text: [1] -
Ishigami, K.; Watanabe, H.; Kitahara, T. Tetrahedron 2005, 61, 7546–7553. doi:10.1016/j.tet.2005.05.057
Return to citation in text: [1] -
Wuts, P. G. M.; Greene, T. W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley and Sons: Hoboken, New Jersey, 2007.
Return to citation in text: [1] -
Hanessian, S.; Focken, T.; Mi, X.; Oza, R.; Chen, B.; Ritson, D.; Beaudegnies, R. J. Org. Chem. 2010, 75, 5601–5618. doi:10.1021/jo100956v
Return to citation in text: [1]
| 37. | Ishigami, K.; Watanabe, H.; Kitahara, T. Tetrahedron 2005, 61, 7546–7553. doi:10.1016/j.tet.2005.05.057 |
| 34. | Hoppen, S.; Bäurle, S.; Koert, U. Chem.–Eur. J. 2000, 6, 2382–2396. doi:10.1002/1521-3765(20000703)6:13<2382::AID-CHEM2382>3.0.CO;2-C |
| 35. | Brandl, T.; Hoffmann, R. W. Eur. J. Org. Chem. 2002, 2613–2623. doi:10.1002/1099-0690(200208)2002:15<2613::AID-EJOC2613>3.0.CO;2-Y |
| 36. | Corey, E. J.; Guzman-Perez, A.; Noe, M. C. Tetrahedron Lett. 1995, 36, 3481–3484. doi:10.1016/0040-4039(95)00570-3 |
| 1. | Kluge, A. F. Heterocycles 1986, 24, 1699–1740. doi:10.3987/R-1986-06-1699 |
| 2. | Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4 |
| 3. | Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015 |
| 4. | Jacobs, M. F.; Kitching, W. B. Curr. Org. Chem. 1998, 2, 395–436. |
| 5. | Francke, W.; Kitching, W. Curr. Org. Chem. 2001, 5, 233–251. doi:10.2174/1385272013375652 |
| 6. | Mead, K. T.; Brewer, B. N. Curr. Org. Chem. 2003, 7, 227–256. doi:10.2174/1385272033372969 |
| 7. | Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n |
| 8. | Raju, B. R.; Saikia, A. K. Molecules 2008, 13, 1942–2038. doi:10.3390/molecules13081942 |
| 9. | Favre, S.; Vogel, P.; Gerber-Lemaire, S. Molecules 2008, 13, 2570–2600. doi:10.3390/molecules13102570 |
| 10. | Sperry, J.; Wilson, Z. E.; Rathwell, D. C. K.; Brimble, M. A. Nat. Prod. Rep. 2010, 27, 1117–1137. doi:10.1039/b911514p |
| 11. | Ballini, R.; Petrini, M.; Rosini, G. Molecules 2008, 13, 319–330. doi:10.3390/molecules13020319 |
| 15. | Mori, K. Bioorg. Med. Chem. 2007, 15, 7505–7523. doi:10.1016/j.bmc.2007.08.040 |
| 33. | Tlais, S. F.; Dudley, G. B. Beilstein J. Org. Chem. 2011, 7, 570–577. doi:10.3762/bjoc.7.66 |
| 13. | Jackson, K. L.; Henderson, J. A.; Motoyoshi, H.; Phillips, A. J. Angew. Chem., Int. Ed. 2009, 48, 2346–2350. doi:10.1002/anie.200806111 |
| 14. | Yamamoto, A.; Ueda, A.; Brémond, P.; Tiseni, P. S.; Kishi, Y. J. Am. Chem. Soc. 2012, 134, 893–896. doi:10.1021/ja2108307 |
| 28. | Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z |
| 7. | Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n |
| 32. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
| 12. | LaCour, T. G.; Guo, C.; Bhandaru, S.; Boyd, M. R.; Fuchs, P. L. J. Am. Chem. Soc. 1998, 120, 692–707. doi:10.1021/ja972160p |
| 32. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
| 22. | Fortner, K. C.; Kato, D.; Tanaka, Y.; Shair, M. D. J. Am. Chem. Soc. 2010, 132, 275–280. doi:10.1021/ja906996c |
| 23. | Phillips, S. T.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 6589–6598. doi:10.1021/ja0705487 |
| 24. | Ravindar, K.; Reddy, M. S.; Lindqvist, L.; Pelletier, J.; Deslongchamps, P. Org. Lett. 2010, 12, 4420–4423. doi:10.1021/ol1019663 |
| 25. | Solladié, G.; Huser, N.; Fischer, J.; Decian, A. J. Org. Chem. 1995, 60, 4988–4990. doi:10.1021/jo00121a015 |
| 28. | Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z |
| 29. | Fernandes, R. A.; Ingle, A. B. Synlett 2010, 158–160. doi:10.1055/s-0029-1218538 |
| 30. | Brimble, M. A.; Finch, O. C.; Heapy, A. M.; Fraser, J. D.; Furkert, D. P.; O’Connor, P. D. Tetrahedron 2011, 67, 995–1001. doi:10.1016/j.tet.2010.11.107 |
| 28. | Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z |
| 29. | Fernandes, R. A.; Ingle, A. B. Synlett 2010, 158–160. doi:10.1055/s-0029-1218538 |
| 30. | Brimble, M. A.; Finch, O. C.; Heapy, A. M.; Fraser, J. D.; Furkert, D. P.; O’Connor, P. D. Tetrahedron 2011, 67, 995–1001. doi:10.1016/j.tet.2010.11.107 |
| 31. | Li, X.; Sattler, I.; Lin, W. J. Antibiot. 2007, 60, 191–195. doi:10.1038/ja.2007.21 |
| 7. | Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n |
| 17. | Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry; Pergamon Press: Oxford, 1983. |
| 18. | Juaristi, E.; Cuevas, G. The Anomeric Effect; CRC Press: Boca Raton, FL, 1994. |
| 19. | Lau, C. K.; Crumpler, S.; Macfarlane, K.; Lee, F.; Berthelette, C. Synlett 2004, 2281–2286. doi:10.1055/s-2004-831334 |
| 20. | Evans, D. A.; Coleman, P. J.; Dias, L. C. Angew. Chem., Int. Ed. Engl. 1997, 36, 2738–2741. doi:10.1002/anie.199727381 |
| 38. | Wuts, P. G. M.; Greene, T. W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley and Sons: Hoboken, New Jersey, 2007. |
| 16. | Byers, J. A.; Högberg, H.-E.; Unelius, C. R.; Birgersson, G.; Löfqvist, J. J. Chem. Ecol. 1989, 15, 685–695. doi:10.1007/BF01014711 |
| 26. | Li, X.; Yao, Y.; Zheng, Y.; Sattler, I.; Lin, W. Arch. Pharmacal Res. 2007, 30, 812–815. doi:10.1007/BF02978829 |
| 27. | Ackland, M. J.; Hanson, J. R.; Hitchcock, P. B.; Ratcliffe, A. H. J. Chem. Soc., Perkin Trans. 1 1985, 843–847. doi:10.1039/P19850000843 |
| 39. | Hanessian, S.; Focken, T.; Mi, X.; Oza, R.; Chen, B.; Ritson, D.; Beaudegnies, R. J. Org. Chem. 2010, 75, 5601–5618. doi:10.1021/jo100956v |
© 2012 Tlais and Dudley; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)