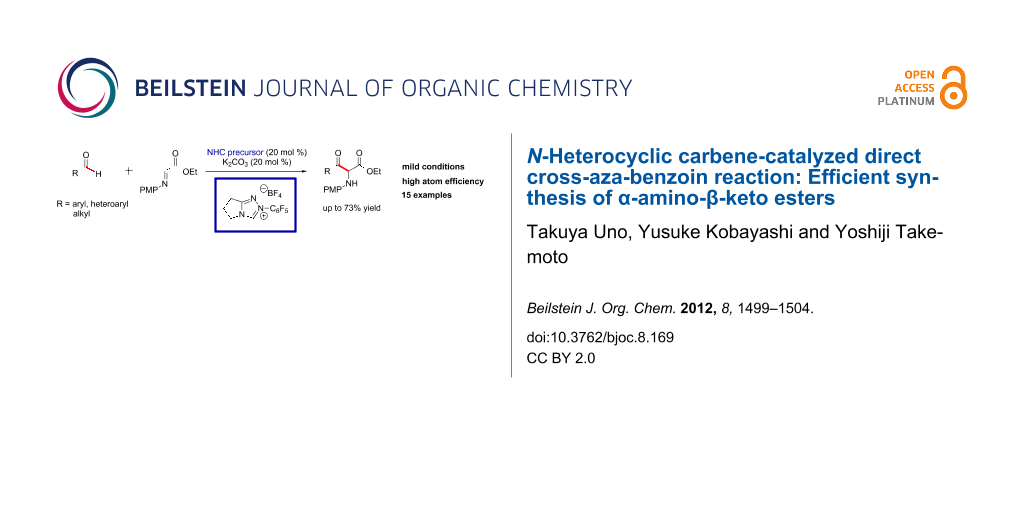
Abstract
An efficient catalytic synthesis of α-amino-β-keto esters has been newly developed. Cross-coupling of various aldehydes with α-imino ester, catalyzed by N-heterocyclic carbene, leads chemoselectively to α-amino-β-keto esters in moderate to good yields with high atom efficiency. The reaction mechanism is discussed, and it is proposed that the α-amino-β-keto esters are formed under thermodynamic control.

Graphical Abstract
Introduction
α-Amino-β-keto ester derivatives are one of the fundamental structural subunits in natural products such as miuraenamides [1], and important building blocks for the synthesis of a variety of heterocyclic compounds [2] and pharmaceutically active products [3-5]. In addition, they are valuable intermediates for chiral α-amino acids [6,7], including β-hydroxy-α-amino acids [8-14]. Consequently, significant efforts have been devoted to synthesizing the privileged structure (Figure 1), and two main distinct approaches based on the bond-forming position have been developed. One approach utilizes Cα–Cβ bond formations, such as (a) acylation of Schiff bases or α-isocyano esters with acyl halides [4,15-17], and (b) intramolecular N–C acyl migration of the N-acyl glycine derivatives [18]. The other consists of C–N bond-forming reactions, such as (c) a rhodium-catalyzed N–H insertion reaction with α-diazo-β-keto esters [19-21], and (d) α-oxidation of β-keto esters to the corresponding oximes and the subsequent hydrogenation [22]. However, the former methods require a stoichiometric amount of strong bases, and the latter employ inaccessible substrates or multistep protocols. Recently, Zhang and co-workers reported Zn(ClO4)2·6H2O-catalyzed, mild and direct α-amination of β-keto esters with TsNH2, but in this case, a stoichiometric amount of PhI=O is needed as the oxidant (Figure 1, (e)) [23]. Therefore, mild, efficient and environmentally friendly strategies for the synthesis of these esters are still needed. We envisioned that highly atom-efficient synthesis of α-amino-β-keto esters could be achieved by a novel umpolung approach including C–C bond construction, that is, formal addition of acyl anion equivalents generated from aldehydes 1 with NHCs, to α-imino esters 2 (Figure 1, (f)).
![[1860-5397-8-169-1]](/bjoc/content/figures/1860-5397-8-169-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthetic methods for α-amino-β-keto esters.
Figure 1: Synthetic methods for α-amino-β-keto esters.
Although a large number of NHC-catalyzed umpolung reactions, such as the benzoin reaction and Stetter reaction, have been developed [24-34], the related reactions of aldehydes with imines, i.e., the cross-aza-benzoin reactions, have been much less studied, in spite of the pharmaceutical and biological importance of providing α-amino ketones [35-43]. In particular, a reaction employing the imines directly has rarely been developed [40,42], due to the unproductive reaction of NHCs with the imines. Therefore, in situ generation of imines or iminium ions from their precursors is generally required to control their reactivity. We anticipated that the introduction of appropriate protecting groups of the nitrogen atom of α-imino esters 2, which have been known to be excellent electrophiles, would suppress an unexpected reaction of 2 with NHCs, enabling the direct use of 2 as acyl anion acceptors in the cross-aza-benzoin reaction. In this communication, we describe a new, efficient, and atom-economical synthesis of the α-amino-β-keto esters by NHC-catalyzed cross-aza-benzoin reaction of aldehydes with α-imino esters under mild basic conditions.
Results and Discussion
First we initiated the cross-aza-benzoin reaction of benzaldehyde (1a) by employing 20 mol % of commercially available precatalyst 3a (Figure 2) and K2CO3. Gratifyingly, when ethyl N-PMP-2-iminoacetate (4) was used as acyl anion acceptor [44], the reaction proceeded smoothly in THF at room temperature to generate the desired product 5a in 58% yield (Table 1, entry 1). Surprisingly, no benzoin 6 arising from homo-coupling of 1a was obtained. Encouraged by this result, we then attempted the other precatalysts 3b–e depicted in Figure 2. Imidazolium salt 3b and simple triazolium salt 3c gave no coupled product 5a (Table 1, entries 2 and 3). Further screening revealed that bicyclic triazolium salt 3d could catalyze the reaction to give 5a in 42% yield (Table 1, entry 4). We reasoned that the acidity of the NHC precursor was important to promote the reaction under mild basic conditions (Table 1, entry 3 versus entry 4), and therefore we next investigated the substituent on the nitrogen atom of NHC. As envisaged, N-pentafluorophenyl-substituted precatalyst 3e [45-49], whose C-2 proton is more acidic than 3d, furnished the coupled product 5a in good yield (Table 1, entry 5). Subsequently, we attempted several bases to find that cesium carbonate gave slightly lower yield (Table 1, entry 6), whereas an amine base and a stronger base were not effective, due to the competitive decomposition of 4 under these conditions (Table 1, entries 7 and 8). Different solvents, such as CH2Cl2, toluene, and MeCN, have been tested in this reaction, and THF was found to be optimal in terms of chemical yield (Table 1, entries 9–11).
![[1860-5397-8-169-2]](/bjoc/content/figures/1860-5397-8-169-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of several NHC precatalysts.
Figure 2: Structures of several NHC precatalysts.
Table 1: Reaction optimization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-169-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cat. 3 | Base | Solvent | Yield (%)b |
|---|---|---|---|---|
| 1 | 3a | K2CO3 | THF | 58 |
| 2 | 3b | K2CO3 | THF | <5 |
| 3 | 3c | K2CO3 | THF | <5 |
| 4 | 3d | K2CO3 | THF | 42 |
| 5 | 3e | K2CO3 | THF | 70 |
| 6 | 3e | Cs2CO3 | THF | 66 |
| 7 | 3e | NEt3 | THF | 56 |
| 8 | 3e | KOt-Bu | THF | 37 |
| 9 | 3e | K2CO3 | CH2Cl2 | 61 |
| 10 | 3e | K2CO3 | toluene | 56 |
| 11 | 3e | K2CO3 | MeCN | 40 |
aReactions conducted with 1a (0.3 mmol) and 4 (1.3 equiv) in THF (0.5 M). bIsolated yields.
With the efficient catalytic system in hand (20 mol % of 3e and K2CO3 in THF at room temperature), we next evaluated the substrate generality with a variety of aromatic aldehydes 1 bearing a diverse range of functionality (Table 2). Chloro-, nitro-, cyano-, and methoxycarbonyl-substituted aromatic aldehydes were compatible with the reaction conditions (Table 2, entries 1–6). In all cases, the reaction led to the desired products in moderate to good yields. In addition, ortho-substituted aldehyde, which is considered to be a poor acyl donor in benzoin reactions [46], was also smoothly converted to the expected product 5d in 61% yield (Table 2, entry 3). It is noteworthy that chloro-substituted aldehydes were tolerated, because the corresponding products 5b–d could in principle undergo further functionalization by palladium-catalyzed cross-coupling reactions. Additionally, electron-rich aromatic aldehydes, which are often known to be less reactive in NHC-catalyzed reactions, could be coupled with 4 to provide 5h and 5i in 66% and 68% yields, respectively (Table 2, entries 7 and 8). Notably, heteroaromatic aldehydes, such as 3-thiophenecarboxaldehyde and furfural, were also successful in yielding the expected products 5j and 5k in 72% and 49% yield, respectively (Table 2, entries 9 and 10).
Table 2: Substrate scope.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-169-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Ar | 5 | Yield (%)b |
|---|---|---|---|
| 1 | 4-ClC6H4 | 5b | 69 |
| 2 | 3-ClC6H4 | 5c | 59 |
| 3 | 2-ClC6H4 | 5d | 61 |
| 4 | 4-NO2C6H4 | 5e | 45 |
| 5 | 4-CNC6H4 | 5f | 39 |
| 6 | 4-MeOCOC6H4 | 5g | 73 |
| 7 | 4-MeC6H4 | 5h | 66 |
| 8 | 4-MeOC6H4 | 5i | 68 |
| 9 | thiophen-3-yl | 5j | 72 |
| 10 | furan-2-yl | 5k | 49 |
aReactions performed with 1 (0.3 mmol) and 4 (1.3 equiv) in THF (0.5 M). bIsolated yields.
We next investigated the reaction with several aliphatic aldehydes (Scheme 1). Generally, unactivated aliphatic aldehydes are unsuccessful for the NHC-catalyzed reactions, because of their low electrophilicity relative to aromatic aldehydes [40,46,47,50]. To our delight, this methodology was found to be also suitable for the enolizable aliphatic aldehydes. Under the optimal reaction conditions (Table 1, entry 5), acetaldehyde and other primary alkyl aldehydes bearing functional groups such as ether, carbamate, and phenyl groups, were converted into the corresponding products 5l–o in good yields. The α-branched aldehyde, however, failed to give the desired product 5p, presumably due to the increased steric hindrance along with the inherently low electrophilicity.
![[1860-5397-8-169-i1]](/bjoc/content/inline/1860-5397-8-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to elucidate the reaction mechanism, a 0.5 equiv of benzoin (6) was used instead of aldehyde 1a under standard conditions, affording the cross-coupled product 5a in 40% yield (Scheme 2). In addition, when the reaction was conducted with α-amino-β-keto ester 5b in place of α-imino ester 4, the cross-over product 5a was not detected. These findings indicate that the formation of benzoin is reversible via the Breslow intermediate, whereas the retro-benzoin reaction of cross-coupled product 5 does not occur under the present reaction conditions, and that the product 5 is formed under thermodynamic control.
![[1860-5397-8-169-i2]](/bjoc/content/inline/1860-5397-8-169-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
A plausible mechanism for the cross-aza-benzoin reaction is shown in Scheme 3. Carbene I is generated by deprotonation of triazolium salt 3e in the presence of K2CO3. The carbene I reacts with aldehyde 1 to afford Breslow intermediate II, which could lead to benzoin (6), or tetrahedral intermediate III when treated with α-imino ester 4. Intermolecular proton transfer from III gives intermediate IV, which could release the product 5 and the carbene I to complete the catalytic system. We speculated that the desired product 5 is thermodynamically more stable than 6 and the formation of 5 is the irreversible step, from the finding that cross-coupled product 5 is predominantly obtained under the reaction conditions.
![[1860-5397-8-169-i3]](/bjoc/content/inline/1860-5397-8-169-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have developed a direct, atom-efficient synthesis of α-amino-β-keto esters by an umpolung reaction. We found that the NHC-catalyzed cross-aza-benzoin reaction of aldehydes with N-PMP-imino ester proceeds chemoselectively under very mild conditions. Therefore, the reaction is tolerant of a range of functional groups and substituents, including aliphatic aldehydes, and thus this method would be an attractive approach for deriving various α-amino-β-keto ester derivatives, which are useful synthetic blocks and valuable pharmaceutical intermediates.
Supporting Information
| Supporting Information File 1: Experimental details and characterization of the synthesized compounds. | ||
| Format: PDF | Size: 424.2 KB | Download |
References
-
Ojika, M.; Inukai, Y.; Kito, Y.; Hirata, M.; Iizuka, T.; Fudou, R. Chem.–Asian J. 2008, 3, 126–133. doi:10.1002/asia.200700233
Return to citation in text: [1] -
Sanz-Cervera, J. F.; Blasco, R.; Piera, J.; Cynamon, M.; Ibáñez, I.; Murguía, M.; Fustero, S. J. Org. Chem. 2009, 74, 8988–8996. doi:10.1021/jo9016265
Return to citation in text: [1] -
Phansavath, P.; Duprat de Paule, S.; Ratovelomanana-Vidal, V.; Genêt, J.-P. Eur. J. Org. Chem. 2000, 3903–3907. doi:10.1002/1099-0690(200012)2000:23<3903::AID-EJOC3903>3.0.CO;2-Q
Return to citation in text: [1] -
Miyake, F.; Hashimoto, M.; Tonseingsom, S.; Yakushijin, K.; Horne, D. A. Tetrahedron 2010, 66, 4888–4893. doi:10.1016/j.tet.2010.03.109
Return to citation in text: [1] [2] -
Wipf, P.; Miller, C. P. J. Org. Chem. 1993, 58, 3604–3606. doi:10.1021/jo00066a004
Return to citation in text: [1] -
Nemoto, T.; Harada, T.; Matsumoto, T.; Hamada, Y. Tetrahedron Lett. 2007, 48, 6304–6307. doi:10.1016/j.tetlet.2007.07.020
Return to citation in text: [1] -
Singh, J.; Gordon, T. D.; Earley, W. G.; Morgan, B. A. Tetrahedron Lett. 1993, 34, 211–214. doi:10.1016/S0040-4039(00)60549-X
Return to citation in text: [1] -
Noyori, R.; Tokunaga, M.; Kitamura, M. Bull. Chem. Soc. Jpn. 1995, 68, 36–55. doi:10.1246/bcsj.68.36
Return to citation in text: [1] -
Makino, K.; Goto, T.; Hiroki, Y.; Hamada, Y. Tetrahedron: Asymmetry 2008, 19, 2816–2828. doi:10.1016/j.tetasy.2008.12.024
Return to citation in text: [1] -
Hamada, Y.; Makino, K. J. Synth. Org. Chem., Jpn. 2008, 66, 1057–1065. doi:10.5059/yukigoseikyokaishi.66.1057
Return to citation in text: [1] -
Liu, Z.; Shultz, C. S.; Sherwood, C. A.; Krska, S.; Dormer, P. G.; Desmond, R.; Lee, C.; Sherer, E. C.; Shpungin, J.; Cuff, J.; Xu, F. Tetrahedron Lett. 2011, 52, 1685–1688. doi:10.1016/j.tetlet.2011.01.146
Return to citation in text: [1] -
Maeda, T.; Makino, K.; Iwasaki, M.; Hamada, Y. Chem.–Eur. J. 2010, 16, 11954–11962. doi:10.1002/chem.201001298
Return to citation in text: [1] -
Seashore-Ludlow, B.; Villo, P.; Häcker, C.; Somfai, P. Org. Lett. 2010, 12, 5274–5277. doi:10.1021/ol102323k
Return to citation in text: [1] -
Hamada, Y.; Koseki, Y.; Fujii, T.; Maeda, T.; Hibino, T.; Makino, K. Chem. Commun. 2008, 6206–6208. doi:10.1039/b816524f
Return to citation in text: [1] -
Suzuki, M.; Iwasaki, T.; Miyoshi, M.; Okumura, K.; Matsumoto, K. J. Org. Chem. 1973, 38, 3571–3575. doi:10.1021/jo00960a028
Return to citation in text: [1] -
Krysan, D. J. Tetrahedron Lett. 1996, 37, 3303–3306. doi:10.1016/0040-4039(96)00581-3
Return to citation in text: [1] -
Schultz, K.; Stief, L.; Kazmaier, U. Synthesis 2012, 600–604. doi:10.1055/s-0031-1289674
Return to citation in text: [1] -
Hara, O.; Ito, M.; Hamada, Y. Tetrahedron Lett. 1998, 39, 5537–5540. doi:10.1016/S0040-4039(98)01093-4
Return to citation in text: [1] -
Bertelsen, S.; Nielsen, M.; Bachmann, S.; Jørgensen, K. A. Synthesis 2005, 13, 2234–2238. doi:10.1055/s-2005-869952
Return to citation in text: [1] -
Bourdon, L. H.; Fairfax, D. J.; Martin, G. S.; Mathison, C. J.; Zhichkin, P. Tetrahedron: Asymmetry 2004, 15, 3485–3487. doi:10.1016/j.tetasy.2004.10.005
Return to citation in text: [1] -
Moody, C. J.; Swann, E. Synlett 1998, 135–136. doi:10.1055/s-1998-1610
Return to citation in text: [1] -
Mordant, C.; Dünkelmann, P.; Ratovelomanana-Vidal, V.; Genet, J.-P. Chem. Commun. 2004, 1296–1297. doi:10.1039/b401631a
Return to citation in text: [1] -
Yu, J.; Liu, S.-S.; Cui, J.; Hou, X.-S.; Zhang, C. Org. Lett. 2012, 14, 832–835. doi:10.1021/ol203358f
Return to citation in text: [1] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380
Return to citation in text: [1] -
Phillips, E. M.; Chan, A.; Scheidt, K. A. Aldrichimica Acta 2009, 42, 55–66.
Return to citation in text: [1] -
Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77–144. doi:10.1007/128_2008_18
Return to citation in text: [1] -
Chiang, P.-C.; Bode, J. W. TCI Mail 2011, 149, 2–17.
Return to citation in text: [1] -
Vora, H. U.; Rovis, T. Aldrichimica Acta 2011, 1, 3–11.
Return to citation in text: [1] -
Hirano, K.; Piel, I.; Glorius, F. Chem. Lett. 2011, 40, 786–791. doi:10.1246/cl.2011.786
Return to citation in text: [1] -
Biju, A. T.; Kuhl, N.; Glorius, F. Acc. Chem. Res. 2011, 44, 1182–1195. doi:10.1021/ar2000716
Return to citation in text: [1] -
Nair, V.; Menon, R. S.; Biju, A. T.; Sinu, C. R.; Paul, R. R.; Jose, A.; Sreekumar, V. Chem. Soc. Rev. 2011, 40, 5336–5346. doi:10.1039/c1cs15139h
Return to citation in text: [1] -
Cohen, D. T.; Scheidt, K. A. Chem. Sci. 2012, 3, 53–57. doi:10.1039/C1SC00621E
Return to citation in text: [1] -
Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e
Return to citation in text: [1] -
Castells, J.; López-Calahorra, F.; Bassedas, M.; Urrios, P. Synthesis 1988, 314–315. doi:10.1055/s-1988-27552
Return to citation in text: [1] -
Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696–9697. doi:10.1021/ja0165943
Return to citation in text: [1] -
Frantz, D. E.; Morency, L.; Soheili, A.; Murry, J. A.; Grabowski, E. J. J.; Tillyer, R. D. Org. Lett. 2004, 6, 843–846. doi:10.1021/ol0498803
Return to citation in text: [1] -
Mattson, A. E.; Scheidt, K. A. Org. Lett. 2004, 6, 4363–4366. doi:10.1021/ol0481129
Return to citation in text: [1] -
Mennen, S. M.; Gipson, J. D.; Kim, Y. R.; Miller, S. J. J. Am. Chem. Soc. 2005, 127, 1654–1655. doi:10.1021/ja042650z
Return to citation in text: [1] -
Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852–854. doi:10.1039/b611646a
Return to citation in text: [1] [2] [3] -
Enders, D.; Henseler, A.; Lowins, S. Synthesis 2009, 24, 4125–4128. doi:10.1055/s-0029-1217070
Return to citation in text: [1] -
DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904–5906. doi:10.1002/anie.201202442
Return to citation in text: [1] [2] -
DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 8094–8097. doi:10.1021/ja3030164
Return to citation in text: [1] -
We employed several imines bearing a variety of substituents on the nitrogen atom (N-Bn, N-t-Bu, N-OBn); however, no desired products were detected.
Return to citation in text: [1] -
Baragwanath, L.; Rose, C. A.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2009, 74, 9214–9217. doi:10.1021/jo902018j
Return to citation in text: [1] -
O'Toole, S. E.; Rose, C. A.; Gundala, S.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2011, 7, 347–357. doi:10.1021/jo101791w
Return to citation in text: [1] [2] [3] -
Jin, M. Y.; Kim, S. M.; Han, H.; Ryu, D. H.; Yang, J. W. Org. Lett. 2011, 13, 880–883. doi:10.1021/ol102937w
Return to citation in text: [1] [2] -
Mao, H.; An, S. L.; Kim, S. M.; Yang, J. W. Bull. Korean Chem. Soc. 2011, 32, 4408–4410. doi:10.5012/bkcs.2011.32.12.4408
Return to citation in text: [1] -
Kim, S. M.; Jin, M. Y.; Kim, M. J.; Cui, Y.; Kim, Y. S.; Zhang, L.; Song, C. E.; Ryu, D. H.; Yang, J. W. Org. Biomol. Chem. 2011, 9, 2069–2071. doi:10.1039/c0ob01178a
Return to citation in text: [1] -
Rose, C. A.; Gundala, S.; Fagan, C.-L.; Franz, J. F.; Connon, S. J.; Zeitler, K. Chem. Sci. 2012, 3, 735–740. doi:10.1039/C2SC00622G
Return to citation in text: [1]
| 40. | Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852–854. doi:10.1039/b611646a |
| 46. | O'Toole, S. E.; Rose, C. A.; Gundala, S.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2011, 7, 347–357. doi:10.1021/jo101791w |
| 47. | Jin, M. Y.; Kim, S. M.; Han, H.; Ryu, D. H.; Yang, J. W. Org. Lett. 2011, 13, 880–883. doi:10.1021/ol102937w |
| 50. | Rose, C. A.; Gundala, S.; Fagan, C.-L.; Franz, J. F.; Connon, S. J.; Zeitler, K. Chem. Sci. 2012, 3, 735–740. doi:10.1039/C2SC00622G |
| 1. | Ojika, M.; Inukai, Y.; Kito, Y.; Hirata, M.; Iizuka, T.; Fudou, R. Chem.–Asian J. 2008, 3, 126–133. doi:10.1002/asia.200700233 |
| 8. | Noyori, R.; Tokunaga, M.; Kitamura, M. Bull. Chem. Soc. Jpn. 1995, 68, 36–55. doi:10.1246/bcsj.68.36 |
| 9. | Makino, K.; Goto, T.; Hiroki, Y.; Hamada, Y. Tetrahedron: Asymmetry 2008, 19, 2816–2828. doi:10.1016/j.tetasy.2008.12.024 |
| 10. | Hamada, Y.; Makino, K. J. Synth. Org. Chem., Jpn. 2008, 66, 1057–1065. doi:10.5059/yukigoseikyokaishi.66.1057 |
| 11. | Liu, Z.; Shultz, C. S.; Sherwood, C. A.; Krska, S.; Dormer, P. G.; Desmond, R.; Lee, C.; Sherer, E. C.; Shpungin, J.; Cuff, J.; Xu, F. Tetrahedron Lett. 2011, 52, 1685–1688. doi:10.1016/j.tetlet.2011.01.146 |
| 12. | Maeda, T.; Makino, K.; Iwasaki, M.; Hamada, Y. Chem.–Eur. J. 2010, 16, 11954–11962. doi:10.1002/chem.201001298 |
| 13. | Seashore-Ludlow, B.; Villo, P.; Häcker, C.; Somfai, P. Org. Lett. 2010, 12, 5274–5277. doi:10.1021/ol102323k |
| 14. | Hamada, Y.; Koseki, Y.; Fujii, T.; Maeda, T.; Hibino, T.; Makino, K. Chem. Commun. 2008, 6206–6208. doi:10.1039/b816524f |
| 45. | Baragwanath, L.; Rose, C. A.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2009, 74, 9214–9217. doi:10.1021/jo902018j |
| 46. | O'Toole, S. E.; Rose, C. A.; Gundala, S.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2011, 7, 347–357. doi:10.1021/jo101791w |
| 47. | Jin, M. Y.; Kim, S. M.; Han, H.; Ryu, D. H.; Yang, J. W. Org. Lett. 2011, 13, 880–883. doi:10.1021/ol102937w |
| 48. | Mao, H.; An, S. L.; Kim, S. M.; Yang, J. W. Bull. Korean Chem. Soc. 2011, 32, 4408–4410. doi:10.5012/bkcs.2011.32.12.4408 |
| 49. | Kim, S. M.; Jin, M. Y.; Kim, M. J.; Cui, Y.; Kim, Y. S.; Zhang, L.; Song, C. E.; Ryu, D. H.; Yang, J. W. Org. Biomol. Chem. 2011, 9, 2069–2071. doi:10.1039/c0ob01178a |
| 6. | Nemoto, T.; Harada, T.; Matsumoto, T.; Hamada, Y. Tetrahedron Lett. 2007, 48, 6304–6307. doi:10.1016/j.tetlet.2007.07.020 |
| 7. | Singh, J.; Gordon, T. D.; Earley, W. G.; Morgan, B. A. Tetrahedron Lett. 1993, 34, 211–214. doi:10.1016/S0040-4039(00)60549-X |
| 46. | O'Toole, S. E.; Rose, C. A.; Gundala, S.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2011, 7, 347–357. doi:10.1021/jo101791w |
| 3. | Phansavath, P.; Duprat de Paule, S.; Ratovelomanana-Vidal, V.; Genêt, J.-P. Eur. J. Org. Chem. 2000, 3903–3907. doi:10.1002/1099-0690(200012)2000:23<3903::AID-EJOC3903>3.0.CO;2-Q |
| 4. | Miyake, F.; Hashimoto, M.; Tonseingsom, S.; Yakushijin, K.; Horne, D. A. Tetrahedron 2010, 66, 4888–4893. doi:10.1016/j.tet.2010.03.109 |
| 5. | Wipf, P.; Miller, C. P. J. Org. Chem. 1993, 58, 3604–3606. doi:10.1021/jo00066a004 |
| 40. | Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852–854. doi:10.1039/b611646a |
| 42. | DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904–5906. doi:10.1002/anie.201202442 |
| 2. | Sanz-Cervera, J. F.; Blasco, R.; Piera, J.; Cynamon, M.; Ibáñez, I.; Murguía, M.; Fustero, S. J. Org. Chem. 2009, 74, 8988–8996. doi:10.1021/jo9016265 |
| 44. | We employed several imines bearing a variety of substituents on the nitrogen atom (N-Bn, N-t-Bu, N-OBn); however, no desired products were detected. |
| 22. | Mordant, C.; Dünkelmann, P.; Ratovelomanana-Vidal, V.; Genet, J.-P. Chem. Commun. 2004, 1296–1297. doi:10.1039/b401631a |
| 24. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 25. | Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380 |
| 26. | Phillips, E. M.; Chan, A.; Scheidt, K. A. Aldrichimica Acta 2009, 42, 55–66. |
| 27. | Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77–144. doi:10.1007/128_2008_18 |
| 28. | Chiang, P.-C.; Bode, J. W. TCI Mail 2011, 149, 2–17. |
| 29. | Vora, H. U.; Rovis, T. Aldrichimica Acta 2011, 1, 3–11. |
| 30. | Hirano, K.; Piel, I.; Glorius, F. Chem. Lett. 2011, 40, 786–791. doi:10.1246/cl.2011.786 |
| 31. | Biju, A. T.; Kuhl, N.; Glorius, F. Acc. Chem. Res. 2011, 44, 1182–1195. doi:10.1021/ar2000716 |
| 32. | Nair, V.; Menon, R. S.; Biju, A. T.; Sinu, C. R.; Paul, R. R.; Jose, A.; Sreekumar, V. Chem. Soc. Rev. 2011, 40, 5336–5346. doi:10.1039/c1cs15139h |
| 33. | Cohen, D. T.; Scheidt, K. A. Chem. Sci. 2012, 3, 53–57. doi:10.1039/C1SC00621E |
| 34. | Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e |
| 19. | Bertelsen, S.; Nielsen, M.; Bachmann, S.; Jørgensen, K. A. Synthesis 2005, 13, 2234–2238. doi:10.1055/s-2005-869952 |
| 20. | Bourdon, L. H.; Fairfax, D. J.; Martin, G. S.; Mathison, C. J.; Zhichkin, P. Tetrahedron: Asymmetry 2004, 15, 3485–3487. doi:10.1016/j.tetasy.2004.10.005 |
| 21. | Moody, C. J.; Swann, E. Synlett 1998, 135–136. doi:10.1055/s-1998-1610 |
| 35. | Castells, J.; López-Calahorra, F.; Bassedas, M.; Urrios, P. Synthesis 1988, 314–315. doi:10.1055/s-1988-27552 |
| 36. | Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696–9697. doi:10.1021/ja0165943 |
| 37. | Frantz, D. E.; Morency, L.; Soheili, A.; Murry, J. A.; Grabowski, E. J. J.; Tillyer, R. D. Org. Lett. 2004, 6, 843–846. doi:10.1021/ol0498803 |
| 38. | Mattson, A. E.; Scheidt, K. A. Org. Lett. 2004, 6, 4363–4366. doi:10.1021/ol0481129 |
| 39. | Mennen, S. M.; Gipson, J. D.; Kim, Y. R.; Miller, S. J. J. Am. Chem. Soc. 2005, 127, 1654–1655. doi:10.1021/ja042650z |
| 40. | Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852–854. doi:10.1039/b611646a |
| 41. | Enders, D.; Henseler, A.; Lowins, S. Synthesis 2009, 24, 4125–4128. doi:10.1055/s-0029-1217070 |
| 42. | DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904–5906. doi:10.1002/anie.201202442 |
| 43. | DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 8094–8097. doi:10.1021/ja3030164 |
| 18. | Hara, O.; Ito, M.; Hamada, Y. Tetrahedron Lett. 1998, 39, 5537–5540. doi:10.1016/S0040-4039(98)01093-4 |
| 4. | Miyake, F.; Hashimoto, M.; Tonseingsom, S.; Yakushijin, K.; Horne, D. A. Tetrahedron 2010, 66, 4888–4893. doi:10.1016/j.tet.2010.03.109 |
| 15. | Suzuki, M.; Iwasaki, T.; Miyoshi, M.; Okumura, K.; Matsumoto, K. J. Org. Chem. 1973, 38, 3571–3575. doi:10.1021/jo00960a028 |
| 16. | Krysan, D. J. Tetrahedron Lett. 1996, 37, 3303–3306. doi:10.1016/0040-4039(96)00581-3 |
| 17. | Schultz, K.; Stief, L.; Kazmaier, U. Synthesis 2012, 600–604. doi:10.1055/s-0031-1289674 |
| 23. | Yu, J.; Liu, S.-S.; Cui, J.; Hou, X.-S.; Zhang, C. Org. Lett. 2012, 14, 832–835. doi:10.1021/ol203358f |
© 2012 Uno et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)