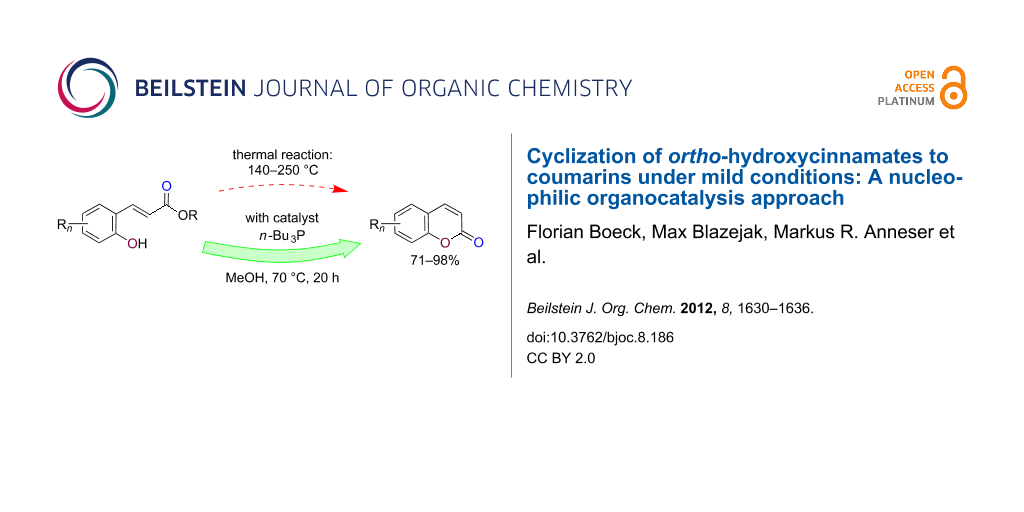
Abstract
(E)-Alkyl ortho-hydroxycinnamates cyclize to coumarins at elevated temperatures of 140–250 °C. We find that the use of tri-n-butylphosphane (20 mol %) as a nucleophilic organocatalyst in MeOH solution allows cyclization to take place under much milder conditions (60–70 °C). Several coumarins were prepared, starting from ortho-hydroxyarylaldehydes, by Wittig reaction with Ph3P=CHCO2Me to (E)-methyl ortho-hydroxycinnamates, followed by the phosphane catalyzed cyclization.

Graphical Abstract
Introduction
Coumarins are important structural motifs in natural products and bioactive compounds, in which they exhibit broad biological activity, e.g., as anticoagulants, antifungal agents, antioxidants, or as anthelmintic, hypnotic and cytotoxic agents [1-4]. Due to their fluorescent properties, coumarins are also widely used as agrochemicals, additives in cosmetics and food, optical brighteners, and dispersed fluorescent and tunable laser-dye optical agents [5,6]. Classical synthetic approaches for coumarins are based on the Perkin reaction or von Pechmann condensation, i.e., reactions under harsh conditions and at elevated temperatures [1]. Recent new methodologies based on CH-activation reactions still use acidic reaction media and show, in part, a lack of regioselectivity [7-10]. Such problems may be circumvented by using ring-closing metathesis [11,12] or other approaches [13-15]. In selective synthetic schemes, the generation of coumarins is typically realized by the cyclization of ortho-hydroxycinnamates (Scheme 1).
![[1860-5397-8-186-i1]](/bjoc/content/inline/1860-5397-8-186-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Conditions for the cyclization of 2’-hydroxycinnamate and related precursors to coumarins. (a) Thermal cyclization [16]. (b) Photochemical isomerization/cyclization [17]. (c) Lewis acid induced demethylation/cyclization [18].
Scheme 1: Conditions for the cyclization of 2’-hydroxycinnamate and related precursors to coumarins. (a) Ther...
This reaction requires high temperatures (140–250 °C, Scheme 1a) [16,19-29] or photochemical double-bond isomerization (Scheme 1b) [15,17,30,31]. An alternative boron tribromide induced lactonization proceeds at a lower temperature (Scheme 1c), but is not compatible with acid-sensitive functionality [18,32]. It follows that synthetic methods converting hydroxycinnamates to coumarins in the absence of acid under mild conditions are very desirable, particularly for labile starting materials, as often found in the late stages of multistep natural-product syntheses.
The difficulty of cyclizing (E)-2’-hydroxycinnamates to coumarins can be traced to the (E)-configuration of the starting material, which places the ester carbonyl group out of reach of the phenolic nucleophile [33-35]. The starting material must first be isomerized to a (Z)-configured intermediate, before cyclization can occur in a geometrically favored manner, but there is a considerable kinetic and energetic barrier against this isomerization process, provoking the observed high reaction temperatures. We wondered whether this problem could be circumvented by adding a nucleophile (HNu) to the reaction mixture containing 1, which is capable of undergoing a reversible conjugate addition to form an intermediate A devoid of an alkene functionality (Scheme 2).
![[1860-5397-8-186-i2]](/bjoc/content/inline/1860-5397-8-186-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Hypothetical catalytic cycle: Nucleophile-assisted cyclization of (E)-ethyl 2’-hydroxycinnamate (1) to coumarin (2).
Scheme 2: Hypothetical catalytic cycle: Nucleophile-assisted cyclization of (E)-ethyl 2’-hydroxycinnamate (1)...
Rotation around the single bond to give B should be a fast process, and cyclization to a 2-chromanone C is then entropically favored. Eventually, the elimination to coumarin (2) could be driven by aromatic stabilization (Scheme 2). In fact, a related stoichiometric two-step protocol has been proposed [36]. It appeared to us that the practical problem of developing a mild and convenient catalytic conversion of ortho-hydroxycinnamates to the corresponding coumarins could be an ideal test case to show the utility of using organocatalytic rationales for solving a synthetic problem.
Results and Discussion
The reaction of (E)-ethyl 2’-hydroxycinnamate (1) to coumarin (2) was chosen as the assay to find catalytic activity under mild conditions (Table 1).
Table 1: Screening of catalysts for nucleophilic double-bond isomerization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-186-i4.svg?max-width=637&scale=1.0)
|
||
| Entry | Catalyst | Yield (%)b |
|---|---|---|
| 1 | PhSH | – |
| 2 | DABCO | – |
| 3 | cinchonine | – |
| 4 | DBU | – |
| 5 | DMAP | – |
| 6 | IMesc | <1 |
| 7 | PPh3 | – |
| 8 | n-Bu3P | 89 |
aReactions were performed on 0.5 mmol scale in 1 mL of solvent; reaction time 20–23 h. bScreening yields were determined by GC and/or HPLC analysis with N-pivaloylaniline as internal standard. cIMes was generated in situ from IMes·HCl [37] and DBU.
It was initially thought that hydro-heteroatomic nucleophiles such as thiols, which are known to easily undergo hetero-Michael additions [38], could be suitable candidates for the screen. However, coumarin was not formed in the presence of thiols (Table 1, entry 1). We turned our attention to nucleophiles that are established catalysts in Morita–Baylis–Hillman type reactions, where they add to conjugated acceptor systems [39]. Tests with either nitrogen bases (Table 1, entries 2–5) or N-heterocyclic carbenes (Table 1, entry 6) were not rewarded with success. Finally, phosphanes were tested, and while triphenylphosphane was not active (Table 1, entry 7), a change to the smaller and more nucleophilic tri-n-butylphosphane, a widely used nucleophilic catalyst [39-42], produced coumarin in rather high yields. Concentrating on n-Bu3P as a very successful catalyst, a solvent screen was performed at the lower temperature of 60 °C (Table 2).
Table 2: Screening of solvents in catalyzed coumarin synthesis.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-186-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Time (h) | Yield (%)b |
|---|---|---|---|
| 1 | toluene | 28 | 26 |
| 2 | isopropanol | 28 | 19 |
| 4 | EtCN | 28 | 8 |
| 5 | dioxane | 28 | 2 |
| 5 | hexane | 20 | 7 |
| 6 | CHCl3 | 28 | 21 |
| 7 | MeOH | 22 | 77 |
| 8 | MeOH | 22 | 47c |
| 9 | glycol | 22 | 3 |
| 10 | glycerol | 16 | 27 |
| 11 | MeOH/H2O (1:1) | 16 | 30 |
| 12 | CF3CH2OH | 16 | 43 |
| 13 | MeCN | 16 | 6 |
| 14 | DMSO | 16 | – |
| 15 | tert-butanol | 23 | 36 |
aReactions were performed on a 0.5 mmol scale in 1 mL of solvent. bScreening yields were determined by GC and/or HPLC analysis with N-pivaloylaniline as the internal standard. cPCy3 was used as the catalyst instead of n-Bu3P.
Conversion to the product in toluene was now considerably reduced (Table 2, entry 1). The broad solvent screening implied that some protic solvents, in particular methanol (Table 2, entry 7), were superior to toluene or polar aprotic solvents. The alternative phosphane PCy3 was also briefly tested in that solvent, but the reduced activity implied that there was a negative steric influence with this catalyst (Table 2, entry 8).The scope and utility of the optimal catalyst system n-Bu3P in MeOH was next studied in preparative reactions with a range of substituted 2’-hydroxycinnamic esters 3, which were readily obtained by reaction of substituted salicylaldehydes with methyl triphenylphosphoranylidene acetate (Ph3P=CHCO2Me) [16,28]. They were cyclized to the corresponding substituted coumarins 4 under the optimized conditions, involving 20 mol % of n-Bu3P as the catalyst in methanol solution at 70 °C (Table 3).
Table 3: Substrate scope of the phosphane catalyzed coumarin synthesis.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-186-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%)b |
|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-186-i7.svg?max-width=637&scale=1.0)
1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-186-i8.svg?max-width=637&scale=1.0)
2 |
82 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-186-i9.svg?max-width=637&scale=1.0)
3a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-186-i10.svg?max-width=637&scale=1.0)
2 |
80 |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-186-i11.svg?max-width=637&scale=1.0)
3b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-186-i12.svg?max-width=637&scale=1.0)
4b |
–c |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-186-i13.svg?max-width=637&scale=1.0)
3c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-186-i14.svg?max-width=637&scale=1.0)
4c |
96 |
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-186-i15.svg?max-width=637&scale=1.0)
3d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-186-i16.svg?max-width=637&scale=1.0)
4d |
88d |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-186-i17.svg?max-width=637&scale=1.0)
3e |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-186-i18.svg?max-width=637&scale=1.0)
4e |
83 |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-186-i19.svg?max-width=637&scale=1.0)
3f |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-186-i20.svg?max-width=637&scale=1.0)
4f |
96 |
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-186-i21.svg?max-width=637&scale=1.0)
3g |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-186-i22.svg?max-width=637&scale=1.0)
4g |
96 |
| 9 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-186-i23.svg?max-width=637&scale=1.0)
3h |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-186-i24.svg?max-width=637&scale=1.0)
4h |
75 |
| 10 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-186-i25.svg?max-width=637&scale=1.0)
3i |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-186-i26.svg?max-width=637&scale=1.0)
4i |
99 |
aReactions were performed on a 1 mmol scale in 1 mL of methanol at 70 °C for 20 h. 15 min prior to workup, the mixture was quenched by addition of 1,2-dibromoethane (20 μL, 0.23 equiv). bYields of pure isolated material. cNo reaction to coumarin observed; starting material (60%) was reisolated. dReaction performed at 90 °C in a closed vessel.
In these preparative experiments, the reaction mixtures were quenched by the addition of a cocatalytic amount of 1,2-dibromoethane prior to work-up, as described below (see the experimental section). The coumarins were generally obtained in good to excellent yields under the satisfactorily mild conditions of this catalysis. An exception was methyl 4’-nitro-2’-hydroxycinnamate, which did not cyclize to coumarin under the present conditions (Table 3, entry 3). Several of the products in Table 3, including the N,N-diethylamino- (Table 3, entry 7), and methoxy-derivatives (Table 3, entries 5 and 6) show strong fluorescence.
The cyclization of (E)-2’-hydroxycinnamates 1 and 3 to coumarins 2 and 4 is slow because it requires prior inversion of the double-bond geometry, a process that calls for harsh conditions such as high temperatures (140–250 °C). We have considered a strategy to perform a fast catalytic conjugate addition of a heteronucleophile to the starting material in order to speed up the overall cyclization reaction. Initial experiments with neutral thiols as nucleophiles were not successful. On the other hand, addition of a nucleophilic trialkylphosphane to (E)-ethyl 2’-hydroxycinnamate (1) in methanol solution, immediately produced a yellow color indicative of the generation of a phenolate anion. When this experiment was carried out in an NMR tube in [D4]-methanol, 31P NMR spectroscopy indicated that n-Bu3P (δ = −30 ppm) was indeed consumed by a conjugate addition to 1, with generation of a tributylphosphonium salt (δ = +37 ppm) of the probable zwitterionic structure 5 (Scheme 3; for the 31P NMR spectrum see the Supporting Information File 1).
![[1860-5397-8-186-i3]](/bjoc/content/inline/1860-5397-8-186-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed catalytic cycle, based on 31P NMR spectroscopic and color evidence.
Scheme 3: Proposed catalytic cycle, based on 31P NMR spectroscopic and color evidence.
Similar phosphonium phenolates are known to exist either as the zwitterionic (analogue to 5) or neutral phosphorane structure (analogue to 5’) [43,44]. Under the conditions of the catalytic reaction, phosphonium (+37 ppm) was the only species of importance and thus represents the resting state of the catalytic reaction, which could be either 5 or 6. We did not observe a 31P NMR signal for n-Bu3P, or a signal for the phosphorane structure 5’, which is not favored in the highly polar solvent methanol [43]. Interestingly, we noted that quenching of the catalytic reaction mixtures with a cocatalytic amount of 1,2-dibromoethane prior to work-up had a favorable effect on product yield and purity: First, any of the catalyst n-Bu3P present will be converted to a water-soluble phosphonium salt, which is easier to separate from the coumarin product than the neutral phosphane is. Second, the yields of coumarins were higher when the quenching procedure was used. This implies that part of the coumarin product may remain associated with the catalyst after full conversion of the starting material, for example in the form of 6 or 6’. Addition of 1,2-dibromoethane will assist in shifting the equilibrium away from 6/6’ to release product 2 and n-Bu3P, since the latter should react irreversibly with the alkylating reagent.
Conclusion
In conclusion, we have developed a new catalytic cyclization of (E)-alkyl 2’-hydroxycinnamates to coumarins with the aid of a nucleophilic alkyl phosphane catalyst, which allows this conversion to be carried out under much milder conditions compared to established procedures [16,19-29]. The reaction should be of particular utility in the generation of coumarin derivatives in the late stages of multistep syntheses of sensitive targets. This work also illustrates the utility of using organocatalytic rationales (mechanistic predictions) for developing new solutions to synthetic problems.
Experimental
General procedure for coumarin synthesis
The starting hydroxycinnamate ester (1 mmol) was inserted in a headspace vial containing a magnetic stirring bar. The vial was flushed with argon and capped. Methanol (1 mL, degassed with argon) was added by syringe through the cap. After addition of tri-n-butylphosphane (50 μL, 0.20 mmol; 20 mol %) with a microliter syringe, the solution turned bright yellow. The reaction mixture was heated to 70 °C and stirred for 20 h. The reaction was quenched by the addition of 1,2-dibromoethane (20 μL, 0.23 mmol, 0.23 equiv) and cooled to room temperature. After evaporation, the crude mixture was purified by column chromatography.
Reaction example: 7-(N,N-Diethylamino)coumarin (4f)
Prepared according to the general procedure from (E)-methyl 3-(2-hydroxy-4-N,N-diethylaminophenyl)propenoate (3f, 249 mg, 1 mmol) with n-Bu3P (50 μL, 0.20 mmol, 20 mol %) in MeOH (1 mL). After work-up and column chromatography (EtOAc/hexanes 1:5 + 5% NEt3), the product was obtained as a yellow crystalline solid (209 mg, 96%). CAS-Nr. 20571-42-0; mp 90 °C; Rf = 0.32 (Hex/EtOAc 5:1 + 5% NEt3); 1H NMR (360 MHz, CDCl3) δ 7.54 (d, J = 9.4 Hz, 1H), 7.25 (d, J = 8.5 Hz, 1H), 6.60 (dd, J = 8.6, 2.6 Hz, 1H), 6.51 (d, J = 2.6 Hz, 1H), 6.04 (d, J = 9.3 Hz, 1H), 3.42 (q, J = 7.1 Hz, 4H), 1.21 (t, J = 7.1 Hz, 1H) ppm; 13C NMR (90 MHz, CDCl3) δ 162.2 (C=O), 156.7 (C), 150.5 (C), 143.7 (CH), 128.7 (CH), 109.3 (CH), 108.9 (CH), 108.5 (C), 97.7 (CH), 45.0 (2 × CH2), 12.4 (2 × CH3) ppm.
See Supporting Information File 1 for experimental details and NMR spectra of coumarins and the precursor 2’-hydroxycinnamates.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 1002.2 KB | Download |
References
-
Sethna, S. M.; Shah, N. M. Chem. Rev. 1945, 36, 1–62. doi:10.1021/cr60113a001
Return to citation in text: [1] [2] -
O’Kennedy, R.; Thomas, R. D., Eds. Coumarins: Biology, Applications, and Mode of Action; Wiley: Chichester, UK, 1997.
Return to citation in text: [1] -
Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S. L.; Lee, K.-H. Med. Res. Rev. 2003, 23, 322–345. doi:10.1002/med.10034
Return to citation in text: [1] -
Murray, R. D. H.; Mendez, J.; Brown, S. A. The Natural Coumarins; Wiley: Chichester, UK, 1982.
Return to citation in text: [1] -
Wagner, B. D. Molecules 2009, 14, 210–231. doi:10.3390/molecules14010210
Return to citation in text: [1] -
Dorlars, A.; Schellhammer, C.-W.; Schroeder, J. Angew. Chem., Int. Ed. Engl. 1975, 14, 665–679. doi:10.1002/anie.197506651
Return to citation in text: [1] -
Jia, C.; Piao, D.; Kitamura, T.; Fujiwara, Y. J. Org. Chem. 2000, 65, 7516–7522. doi:10.1021/jo000861q
Return to citation in text: [1] -
Kitamura, T.; Yamamoto, K.; Kotani, M.; Oyamada, J.; Jia, C.; Fujiwara, Y. Bull. Chem. Soc. Jpn. 2003, 76, 1889–1895. doi:10.1246/bcsj.76.1889
Return to citation in text: [1] -
Kotani, M.; Yamamoto, K.; Oyamada, J.; Fujiwara, Y.; Kitamura, T. A. Synthesis 2004, 1466–1470. doi:10.1055/s-2004-822360
Return to citation in text: [1] -
Wegner, H. A.; Ahles, S.; Neuburger, M. Chem.–Eur. J. 2010, 14, 11310–11313. doi:10.1002/chem.200801848
Return to citation in text: [1] -
Chatterjee, A. K.; Toste, F. D.; Goldberg, S. D.; Grubbs, R. H. Pure Appl. Chem. 2003, 75, 421–425. doi:10.1351/pac200375040421
Return to citation in text: [1] -
Schmidt, B.; Krehl, S. Chem. Commun. 2011, 47, 5879–5881. doi:10.1039/c1cc11347j
Return to citation in text: [1] -
Gross, U.; Gross, P. J.; Shi, M.; Bräse, S. Synlett 2011, 635–638. doi:10.1055/s-0030-1259691
Return to citation in text: [1] -
Zeitler, K.; Rose, C. A. J. Org. Chem. 2009, 74, 1759–1762. doi:10.1021/jo802285r
Return to citation in text: [1] -
Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Masuno, H.; Kinoshita, M.; Kagechika, H.; Tanatani, A. J. Med. Chem. 2011, 54, 7055–7065. doi:10.1021/jm2005404
Return to citation in text: [1] [2] -
Narasimhan, N. S.; Mali, R. S.; Barve, M. V. Synthesis 1979, 906–909. doi:10.1055/s-1979-28871
Return to citation in text: [1] [2] [3] [4] -
Horaguchi, T.; Hosokawa, N.; Tanemura, K.; Suzuki, T. J. Heterocycl. Chem. 2002, 39, 61–67. doi:10.1002/jhet.5570390108
Return to citation in text: [1] [2] -
Cairns, N.; Harwood, L. M.; Astles, D. P.; Orr, A. J. Chem. Soc., Chem. Commun. 1986, 182–183. doi:10.1039/C39860000182
Return to citation in text: [1] [2] -
Guillaumet, G.; Hretani, M.; Coudert, G. Tetrahedron Lett. 1988, 29, 2665–2666. doi:10.1016/0040-4039(88)85254-7
Return to citation in text: [1] [2] -
Ishii, H.; Kaneko, Y.; Miyazaki, H.; Harayama, T. Chem. Pharm. Bull. 1991, 39, 3100–3102. doi:10.1248/cpb.39.3100
Return to citation in text: [1] [2] -
Zubia, E.; Rodriguez Luis, F.; Massanet, G. M.; Collado, I. G. Tetrahedron 1992, 48, 4239–4246. doi:10.1016/S0040-4020(01)92200-8
Return to citation in text: [1] [2] -
Harayama, T.; Katsuno, K.; Nishioka, H.; Fujii, M.; Nishita, Y.; Ishii, H.; Kaneko, Y. Heterocycles 1994, 39, 613–622. doi:10.3987/COM-94-S(B)54
Return to citation in text: [1] [2] -
Besson, T.; Ruiz, N.; Coudert, G.; Guillaumet, G. Tetrahedron 1995, 51, 3197–3204. doi:10.1016/0040-4020(95)00058-G
Return to citation in text: [1] [2] -
Cartwright, G. A.; McNab, H. J. Chem. Res., Synop. 1997, 296–297. doi:10.1039/A702405C
Return to citation in text: [1] [2] -
Park, S. B.; Alper, H. Org. Lett. 2003, 5, 3209–3212. doi:10.1021/ol030071d
Return to citation in text: [1] [2] -
Audisio, D.; Messaoudi, S.; Brion, J.-D.; Alami, M. Eur. J. Org. Chem. 2010, 1046–1051. doi:10.1002/ejoc.200901107
Return to citation in text: [1] [2] -
Oberholzer, M.; Gerber, R.; Frech, C. M. Adv. Synth. Catal. 2012, 354, 627–641. doi:10.1002/adsc.201100646
Return to citation in text: [1] [2] -
Mali, R. S.; Yadav, V. J. Synthesis 1977, 464–465. doi:10.1055/s-1977-24443
Return to citation in text: [1] [2] [3] -
Mali, R. S.; Tilve, S. G.; Yeola, S. N.; Manekar, A. R. Heterocycles 1987, 26, 121–127. doi:10.3987/R-1987-01-0121
Return to citation in text: [1] [2] -
Stoermer, R. Ber. Dtsch. Chem. Ges. 1911, 44, 637–668. doi:10.1002/cber.19110440194
Return to citation in text: [1] -
Stoermer, R. Ber. Dtsch. Chem. Ges. 1909, 42, 4865–4871. doi:10.1002/cber.190904204108
Return to citation in text: [1] -
Dubuffet, T.; Luotz, A.; Lavielle, G. Synth. Commun. 1999, 29, 929–936. doi:10.1080/00397919908086054
Return to citation in text: [1] -
Hershfield, R.; Schmir, G. L. J. Am. Chem. Soc. 1973, 95, 7359–7369. doi:10.1021/ja00803a025
Return to citation in text: [1] -
Porter, N. A.; Bruhnke, J. D. J. Am. Chem. Soc. 1989, 111, 7616–7618. doi:10.1021/ja00201a054
Return to citation in text: [1] -
Gagey, N.; Neveu, P.; Benbrahim, C.; Goetz, B.; Aujard, I.; Baudin, J.-B.; Jullien, L. J. Am. Chem. Soc. 2007, 129, 9986–9998. doi:10.1021/ja0722022
Return to citation in text: [1] -
Mukerjee, A. K.; Joseph, K.; Rao, L. J. Chem. Soc., Perkin Trans. 1 1991, 2611–2612. doi:10.1039/P19910002611
Return to citation in text: [1] -
Hintermann, L. Beilstein J. Org. Chem. 2007, 3, No. 22. doi:10.1186/1860-5397-3-22
Return to citation in text: [1] -
Perlmutter, P. Conjugate addition reactions in organic synthesis; Pergamon Press: Oxford, 1992.
Return to citation in text: [1] -
Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–891. doi:10.1021/cr010043d
Return to citation in text: [1] [2] -
Gimbert, C.; Moreno-Mañas, M.; Pérez, E.; Vallribera, A. Tetrahedron 2007, 63, 8305–8310. doi:10.1016/j.tet.2007.05.088
Return to citation in text: [1] -
Webber, P. A. Studies Toward the Total Synthesis of Quinine. Ph.D. Thesis, University of Texas, Austin, 2008.
Return to citation in text: [1] -
Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087
Return to citation in text: [1] -
Begasse, B.; Le Corre, M. Tetrahedron 1980, 36, 3409–3412. doi:10.1016/0040-4020(80)80192-X
Return to citation in text: [1] [2] -
Zhu, X.-F.; Henry, C. E.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 6722–6723. doi:10.1021/ja071990s
Return to citation in text: [1]
| 16. | Narasimhan, N. S.; Mali, R. S.; Barve, M. V. Synthesis 1979, 906–909. doi:10.1055/s-1979-28871 |
| 28. | Mali, R. S.; Yadav, V. J. Synthesis 1977, 464–465. doi:10.1055/s-1977-24443 |
| 39. | Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–891. doi:10.1021/cr010043d |
| 39. | Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–891. doi:10.1021/cr010043d |
| 40. | Gimbert, C.; Moreno-Mañas, M.; Pérez, E.; Vallribera, A. Tetrahedron 2007, 63, 8305–8310. doi:10.1016/j.tet.2007.05.088 |
| 41. | Webber, P. A. Studies Toward the Total Synthesis of Quinine. Ph.D. Thesis, University of Texas, Austin, 2008. |
| 42. | Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087 |
| 1. | Sethna, S. M.; Shah, N. M. Chem. Rev. 1945, 36, 1–62. doi:10.1021/cr60113a001 |
| 2. | O’Kennedy, R.; Thomas, R. D., Eds. Coumarins: Biology, Applications, and Mode of Action; Wiley: Chichester, UK, 1997. |
| 3. | Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S. L.; Lee, K.-H. Med. Res. Rev. 2003, 23, 322–345. doi:10.1002/med.10034 |
| 4. | Murray, R. D. H.; Mendez, J.; Brown, S. A. The Natural Coumarins; Wiley: Chichester, UK, 1982. |
| 11. | Chatterjee, A. K.; Toste, F. D.; Goldberg, S. D.; Grubbs, R. H. Pure Appl. Chem. 2003, 75, 421–425. doi:10.1351/pac200375040421 |
| 12. | Schmidt, B.; Krehl, S. Chem. Commun. 2011, 47, 5879–5881. doi:10.1039/c1cc11347j |
| 37. | Hintermann, L. Beilstein J. Org. Chem. 2007, 3, No. 22. doi:10.1186/1860-5397-3-22 |
| 7. | Jia, C.; Piao, D.; Kitamura, T.; Fujiwara, Y. J. Org. Chem. 2000, 65, 7516–7522. doi:10.1021/jo000861q |
| 8. | Kitamura, T.; Yamamoto, K.; Kotani, M.; Oyamada, J.; Jia, C.; Fujiwara, Y. Bull. Chem. Soc. Jpn. 2003, 76, 1889–1895. doi:10.1246/bcsj.76.1889 |
| 9. | Kotani, M.; Yamamoto, K.; Oyamada, J.; Fujiwara, Y.; Kitamura, T. A. Synthesis 2004, 1466–1470. doi:10.1055/s-2004-822360 |
| 10. | Wegner, H. A.; Ahles, S.; Neuburger, M. Chem.–Eur. J. 2010, 14, 11310–11313. doi:10.1002/chem.200801848 |
| 38. | Perlmutter, P. Conjugate addition reactions in organic synthesis; Pergamon Press: Oxford, 1992. |
| 33. | Hershfield, R.; Schmir, G. L. J. Am. Chem. Soc. 1973, 95, 7359–7369. doi:10.1021/ja00803a025 |
| 34. | Porter, N. A.; Bruhnke, J. D. J. Am. Chem. Soc. 1989, 111, 7616–7618. doi:10.1021/ja00201a054 |
| 35. | Gagey, N.; Neveu, P.; Benbrahim, C.; Goetz, B.; Aujard, I.; Baudin, J.-B.; Jullien, L. J. Am. Chem. Soc. 2007, 129, 9986–9998. doi:10.1021/ja0722022 |
| 5. | Wagner, B. D. Molecules 2009, 14, 210–231. doi:10.3390/molecules14010210 |
| 6. | Dorlars, A.; Schellhammer, C.-W.; Schroeder, J. Angew. Chem., Int. Ed. Engl. 1975, 14, 665–679. doi:10.1002/anie.197506651 |
| 36. | Mukerjee, A. K.; Joseph, K.; Rao, L. J. Chem. Soc., Perkin Trans. 1 1991, 2611–2612. doi:10.1039/P19910002611 |
| 18. | Cairns, N.; Harwood, L. M.; Astles, D. P.; Orr, A. J. Chem. Soc., Chem. Commun. 1986, 182–183. doi:10.1039/C39860000182 |
| 15. | Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Masuno, H.; Kinoshita, M.; Kagechika, H.; Tanatani, A. J. Med. Chem. 2011, 54, 7055–7065. doi:10.1021/jm2005404 |
| 17. | Horaguchi, T.; Hosokawa, N.; Tanemura, K.; Suzuki, T. J. Heterocycl. Chem. 2002, 39, 61–67. doi:10.1002/jhet.5570390108 |
| 30. | Stoermer, R. Ber. Dtsch. Chem. Ges. 1911, 44, 637–668. doi:10.1002/cber.19110440194 |
| 31. | Stoermer, R. Ber. Dtsch. Chem. Ges. 1909, 42, 4865–4871. doi:10.1002/cber.190904204108 |
| 16. | Narasimhan, N. S.; Mali, R. S.; Barve, M. V. Synthesis 1979, 906–909. doi:10.1055/s-1979-28871 |
| 19. | Guillaumet, G.; Hretani, M.; Coudert, G. Tetrahedron Lett. 1988, 29, 2665–2666. doi:10.1016/0040-4039(88)85254-7 |
| 20. | Ishii, H.; Kaneko, Y.; Miyazaki, H.; Harayama, T. Chem. Pharm. Bull. 1991, 39, 3100–3102. doi:10.1248/cpb.39.3100 |
| 21. | Zubia, E.; Rodriguez Luis, F.; Massanet, G. M.; Collado, I. G. Tetrahedron 1992, 48, 4239–4246. doi:10.1016/S0040-4020(01)92200-8 |
| 22. | Harayama, T.; Katsuno, K.; Nishioka, H.; Fujii, M.; Nishita, Y.; Ishii, H.; Kaneko, Y. Heterocycles 1994, 39, 613–622. doi:10.3987/COM-94-S(B)54 |
| 23. | Besson, T.; Ruiz, N.; Coudert, G.; Guillaumet, G. Tetrahedron 1995, 51, 3197–3204. doi:10.1016/0040-4020(95)00058-G |
| 24. | Cartwright, G. A.; McNab, H. J. Chem. Res., Synop. 1997, 296–297. doi:10.1039/A702405C |
| 25. | Park, S. B.; Alper, H. Org. Lett. 2003, 5, 3209–3212. doi:10.1021/ol030071d |
| 26. | Audisio, D.; Messaoudi, S.; Brion, J.-D.; Alami, M. Eur. J. Org. Chem. 2010, 1046–1051. doi:10.1002/ejoc.200901107 |
| 27. | Oberholzer, M.; Gerber, R.; Frech, C. M. Adv. Synth. Catal. 2012, 354, 627–641. doi:10.1002/adsc.201100646 |
| 28. | Mali, R. S.; Yadav, V. J. Synthesis 1977, 464–465. doi:10.1055/s-1977-24443 |
| 29. | Mali, R. S.; Tilve, S. G.; Yeola, S. N.; Manekar, A. R. Heterocycles 1987, 26, 121–127. doi:10.3987/R-1987-01-0121 |
| 17. | Horaguchi, T.; Hosokawa, N.; Tanemura, K.; Suzuki, T. J. Heterocycl. Chem. 2002, 39, 61–67. doi:10.1002/jhet.5570390108 |
| 18. | Cairns, N.; Harwood, L. M.; Astles, D. P.; Orr, A. J. Chem. Soc., Chem. Commun. 1986, 182–183. doi:10.1039/C39860000182 |
| 32. | Dubuffet, T.; Luotz, A.; Lavielle, G. Synth. Commun. 1999, 29, 929–936. doi:10.1080/00397919908086054 |
| 16. | Narasimhan, N. S.; Mali, R. S.; Barve, M. V. Synthesis 1979, 906–909. doi:10.1055/s-1979-28871 |
| 43. | Begasse, B.; Le Corre, M. Tetrahedron 1980, 36, 3409–3412. doi:10.1016/0040-4020(80)80192-X |
| 44. | Zhu, X.-F.; Henry, C. E.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 6722–6723. doi:10.1021/ja071990s |
| 13. | Gross, U.; Gross, P. J.; Shi, M.; Bräse, S. Synlett 2011, 635–638. doi:10.1055/s-0030-1259691 |
| 14. | Zeitler, K.; Rose, C. A. J. Org. Chem. 2009, 74, 1759–1762. doi:10.1021/jo802285r |
| 15. | Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Masuno, H.; Kinoshita, M.; Kagechika, H.; Tanatani, A. J. Med. Chem. 2011, 54, 7055–7065. doi:10.1021/jm2005404 |
| 16. | Narasimhan, N. S.; Mali, R. S.; Barve, M. V. Synthesis 1979, 906–909. doi:10.1055/s-1979-28871 |
| 19. | Guillaumet, G.; Hretani, M.; Coudert, G. Tetrahedron Lett. 1988, 29, 2665–2666. doi:10.1016/0040-4039(88)85254-7 |
| 20. | Ishii, H.; Kaneko, Y.; Miyazaki, H.; Harayama, T. Chem. Pharm. Bull. 1991, 39, 3100–3102. doi:10.1248/cpb.39.3100 |
| 21. | Zubia, E.; Rodriguez Luis, F.; Massanet, G. M.; Collado, I. G. Tetrahedron 1992, 48, 4239–4246. doi:10.1016/S0040-4020(01)92200-8 |
| 22. | Harayama, T.; Katsuno, K.; Nishioka, H.; Fujii, M.; Nishita, Y.; Ishii, H.; Kaneko, Y. Heterocycles 1994, 39, 613–622. doi:10.3987/COM-94-S(B)54 |
| 23. | Besson, T.; Ruiz, N.; Coudert, G.; Guillaumet, G. Tetrahedron 1995, 51, 3197–3204. doi:10.1016/0040-4020(95)00058-G |
| 24. | Cartwright, G. A.; McNab, H. J. Chem. Res., Synop. 1997, 296–297. doi:10.1039/A702405C |
| 25. | Park, S. B.; Alper, H. Org. Lett. 2003, 5, 3209–3212. doi:10.1021/ol030071d |
| 26. | Audisio, D.; Messaoudi, S.; Brion, J.-D.; Alami, M. Eur. J. Org. Chem. 2010, 1046–1051. doi:10.1002/ejoc.200901107 |
| 27. | Oberholzer, M.; Gerber, R.; Frech, C. M. Adv. Synth. Catal. 2012, 354, 627–641. doi:10.1002/adsc.201100646 |
| 28. | Mali, R. S.; Yadav, V. J. Synthesis 1977, 464–465. doi:10.1055/s-1977-24443 |
| 29. | Mali, R. S.; Tilve, S. G.; Yeola, S. N.; Manekar, A. R. Heterocycles 1987, 26, 121–127. doi:10.3987/R-1987-01-0121 |
| 43. | Begasse, B.; Le Corre, M. Tetrahedron 1980, 36, 3409–3412. doi:10.1016/0040-4020(80)80192-X |
© 2012 Boeck et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)