Abstract
In this work we developed C2-symmetric chiral nucleophilic catalysts which possess a pyrrolidinopyridine framework as a catalytic site. Some of these organocatalysts effectively promoted asymmetric desymmetrization of meso-diols via enantioselective acylation.

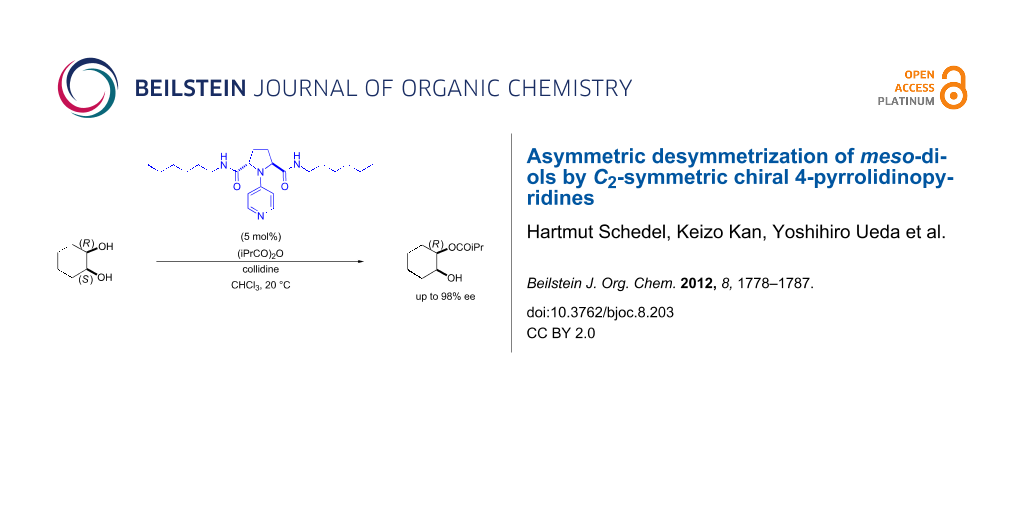
Graphical Abstract
Introduction
Since the pioneering discovery of a catalyst for enantioselective acylation by Vedejs [1], numerous efforts have been devoted to the development of catalysts for enantioselective acylation [2,3]. We have focused on the development of chiral nucleophilic catalysts possessing a pyrrolidinopyridine (PPY) framework as a catalytic site because PPY has been known to be one of the most powerful catalysts for the acylation of alcohols [4-7]. The salient feature of our catalyst design is to introduce chiral elements far from the catalytically active pyridine nitrogen as shown in Figure 1 [8-18]. These catalysts are expected to show high catalytic activity because the introduction of substituents close to the pyridine nitrogen has been known to result in the significant decrease of the catalytic activity [19]. Catalyst 1 was demonstrated to be effective for the kinetic resolution of racemic diols (s: up to 12) [8] and amino alcohol derivatives (s: up to 54) [9]. Catalyst 2, readily prepared from L-proline, could be employed for the kinetic resolution of amino alcohol derivatives (s: up to 11) [10]. Chiral PPY catalysts with dual functional side chains at C(2) and C(4) of the pyrrolidine ring such as 3 were prepared from trans-4-hydroxy-L-proline. These catalysts were found to be moderately effective for the asymmetric desymmetrization of meso-diols [11]. C2-Symmetric PPY-catalyst 4 was found to be effective for the chemo- and regioselective acylation of carbohydrates [12,14,16] and the chemoselective monoacylation of linear diols [17]. Here, we report the asymmetric desymmtrization of meso-diols by C2-symmetric PPY catalysts [20]. The effects of the functional side chains at C(2) and C(5) on the efficiency of the asymmetric desymmetrization are discussed. Some of the results shown here have already been appeared in the patent JP2005132746.
![[1860-5397-8-203-1]](/bjoc/content/figures/1860-5397-8-203-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Asymmetric desymmetrization of meso-1,2-cyclohexanediol
The asymmetric desymmetrization of meso-diols via organocatalytic enantioselective acylation has been extensively studied [1,21-38]. We have reported the asymmetric desymmetrization of meso-1,2-cyclohexanediol (5) by catalytic enantioselective acylation with catalyst 3 (Scheme 1) [11]. Among various chiral PPY catalysts with dual functional side chains at C(2) and C(4) of the pyrrolidine ring, catalyst 3 was found to be most effective for the asymmetric desymmetrization (Scheme 1). However, the enantioselectivity of the asymmetric desymmetrization was far from being sufficient. Molecular modeling of the related catalysts indicated that a C2-symmetric PPY catalyst with functional side chains at C(2) and C(5) might be better suited for this purpose [12,13]. Based on these background, we have prepared various C2-symmetric chiral PPY-catalysts according to Scheme 2 [12] and employed them for asymmetric desymmetrization of meso-1,2-cyclohexanediol (5) [20].
![[1860-5397-8-203-i1]](/bjoc/content/inline/1860-5397-8-203-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric desymmetrization of 5 with catalyst 3.
Scheme 1: Asymmetric desymmetrization of 5 with catalyst 3.
![[1860-5397-8-203-i2]](/bjoc/content/inline/1860-5397-8-203-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of a small library of chiral C2-symmetric PPY catalysts (reference, see [12]).
Scheme 2: Preparation of a small library of chiral C2-symmetric PPY catalysts (reference, see [12]).
We first examined chiral PPY catalyst 8a with an L-tryptophan side chain, which was disclosed to be an excellent catalyst for the regioselective acylation of glycopyranoses [12]. Reaction of 5 with isobutyric anhydride (1.3 equiv) in the presence of 5 mol % of 8a in chloroform at 20 °C gave monoacylate 6 in 73% ee and 85% yield with concomitant formation of 15% of diacylate 7 (Table 1, entry 1). Catalyst 8b with a D-tryptophan side chain gave a lower enantioselectivity (54% ee, Table 1, entry 2). The hydroxy group at the (R)-chiral center was preferentially acylated in both cases. Since both catalysts 8a with an L-tryptophan side chain and 8b with a D-tryptophan side chain gave (1R,2S)-6 [8] by asymmetric desymmetrization of 5, catalysts with achiral side chains were then examined. The acylation of 5 with catalyst 9 which possesses a tryptamine moiety gave 6 in 72% ee and 74% yield (Table 1, entry 3). Catalyst 10 with a glycine moiety gave 6 in 71% ee and 72% yield on treatment of 5 (Table 1, entry 4). Monoacylate 6 was also obtained in 87% ee and 75% yield by acylation of 5 with catalyst 11 which possesses a simple n-hexyl side chain (Table 1, entry 5). Catalysts with chiral side chains, 12a, 12b, 13, and 14, possessing L-β-phenylalanine, D-β-phenylalanine, L-valine, or L-leucine moiety, respectively, also gave 6 in 54–83% ee and 70–77% yields via acylative asymmetric desymmetrization of 5 with isobutyric anhydride (Table 1, entries 6–9). In each case, (1R,2S)-6 was preferentially obtained. These results indicate that the functionality and chirality of catalyst side chains do not affect the absolute configuration of the monoacylate obtained by the asymmetric desymmetrization while they influence the extent of the enantioselectivity. Accordingly, the configuration of the stereocenters in C(2) and C(5) position bearing the amide substituents appears to have decisive effects on the stereochemical course of the asymmetric desymmetrization.
Table 1: Effects of the catalysts’ side chains on the asymmetric desymmetrization of meso-1,2-cyclohexanediol (5).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-203-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalystb | 6:7:Recovery of 5 (%)c | ee of 6 (%)d,e |
|---|---|---|---|
| 1 | 8a | 85:15:0 | 73 |
| 2 | 8b | 70:18:12 | 54 |
| 3 | 9 | 74:16:10 | 72 |
| 4 | 10 | 72:19:9 | 71 |
| 5 | 11 | 75:23:3 | 87 |
| 6 | 12a | 70:25:5 | 74 |
| 7 | 12b | 77:14:9 | 83 |
| 8 | 13 | 76:18:6 | 54 |
| 9 | 14 | 75:20:5 | 81 |
aReactions were run at a substrate concentration of 0.2 M.
bStructures of catalysts:
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-203-i6.svg?max-width=637&scale=1.18182) .
.
cYields determined by 1H NMR with dibenzyl ether as an internal standard. dDetermined by GC analysis with a chiral stationary phase, beta-DEX 225. e(1R,2S)-Isomer was obtained in each case.
Since catalyst 12b showed relatively high enantioselectivity (83% ee) and mono/diacylation ratio (77:14), we next investigated the solvent effects of the asymmetric desymmetrization of 5 with isobutyric anhydride in the presence of catalyst 12b (Table 2, entries 1–5). A clear relationship between the enantioselectivity and the solvent polarity was observed: The lower the polarity of the solvent, the higher the enantioselectivity. This suggests that the hydrogen-bonding interaction between the catalyst and the substrate may be involved in the transition state of the enantioselective acylation (Figure 2 and Figure 3). The temperature effects (lower temperature) of the asymmetric desymmetrization employing catalysts 14 and 11 were examined because both of the catalysts show high solubility in chloroform at low temperatures (Table 2, entries 6–9). Both enantioselectivity (87% ee) and mono/diacylation ratio (85:6) of the acylation of 5 catalyzed by 14 at −40 °C were improved compared with those in the corresponding reaction at 20 °C (Table 2, entry 6 vs entry 7). Similarly, the efficiency of the asymmetric desymmetrization of 5 catalyzed by 11 was improved by conducting the reaction at −40 °C (Table 2, entry 8 vs entry 9). Monoacylate 6 was obtained in 88% ee and 92% yield by treatment of 5 with isobutyric anhydride in the presence of 5 mol % of 11 at −40 °C.
Table 2: Effects of solvents and temperature on the asymmetric desymmetrization of 5.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-203-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Solvent | Temp. (°C) | 6:7:recovery of 5 (%)b | ee of 6 (%)c,d |
|---|---|---|---|---|---|
| 1 | 12b | CCl4 | 20 °C | 75:20:5 | 93 |
| 2 | 12b | toluene | 20 °C | 75:19:6 | 91 |
| 3 | 12b | CHCl3 | 20 °C | 77:14:9 | 83 |
| 4 | 12b | THF | 20 °C | 57:28:15 | 51 |
| 5 | 12b | CH3CN | 20 °C | 69:23:8 | 34 |
| 6 | 14 | CHCl3 | 20 °C | 75:20:5 | 81 |
| 7e | 14 | CHCl3 | −40 °C | 85:6:9 | 87 |
| 8 | 11 | CHCl3 | 20 °C | 75:23:2 | 87 |
| 9e | 11 | CHCl3 | −40 °C | 92:5:3 | 88 |
aReactions were run at a substrate concentration of 0.2 M. bYields determined by 1H NMR with dibenzyl ether as an internal standard. cDetermined by GC analysis with a chiral stationary phase, beta-DEX 225. d(1R,2S)-Isomer was obtained in each case. eRun for 24 h.
Further optimization of the asymmetric desymmetrization of 5 with catalyst 11 was examined at 20 °C (Table 3). The use of only 0.5 mol % of catalyst was found to be still effective in the asymmetric desymmetrization of 5 to give 6 in 90% ee and 76% yield (Table 3, entry 2) [20]. Further decrease in the amount of the catalyst to 0.05 mol % resulted in a lower enantioselectivity (74% ee) and in lower yield (66%) (Table 3, entry 3). The use of a less amount (1.0 equiv) of the anhydride in the presence of 5 mol % of 11 improved the mono/diacylation ratio (84:7), while the enantioselectivity was decreased (81% ee, Table 3, entry 1 vs entry 4). On the other hand, the use of an excess amount (1.6 equiv) of the anhydride resulted in the highest enantioselectivity (98% ee) in compensation for the low yield (59%) for monoacylation (Table 3, entry 5) [20]. The increase in the amount of diacylate 7 is associated with the higher ee of monoacylate 6 (Table 3, entries 1, 4 and 5). This suggests that the ee of monoacylate 6 would be amplified by the second acylation step, i.e., acylative kinetic resolution of enantioenriched monoacylate 6 produced by the asymmetric desymmetrization of the meso-substrate (Scheme 3).
Table 3: Optimization of the asymmetric desymmetrization of 5 with catalyst 11.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-203-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | Mol % of 11 | Equiv of (iPrCO)2O | 6:7:Recovery of 5 (%)b | ee of 6 (%)c,d |
|---|---|---|---|---|
| 1 | 5 | 1.3 | 75:23:2 | 87 |
| 2e | 0.5 | 1.3 | 76:20:3 | 90 |
| 3 | 0.05 | 1.3 | 66:10:24 | 74 |
| 4 | 5 | 1.0 | 84: 7:4 | 81 |
| 5e,f | 5 | 1.6 | 59:41:0 | 98 |
aReactions were run at a substrate concentration of 0.2 M. bYields determined by 1H NMR with dibenzyl ether as an internal standard. cDetermined by GC analysis with a chiral stationary phase, beta-DEX 225. d(1R,2S)-Isomer was obtained in each case. eData quoted from reference [20]. f1.7 Equiv of collidine were used.
![[1860-5397-8-203-i3]](/bjoc/content/inline/1860-5397-8-203-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Amplification of enantiomeric purity of the major enantiomer produced at the step of asymmetric desymmetrization of the meso-substrate by the following kinetic resolution with the same catalyst.
Scheme 3: Amplification of enantiomeric purity of the major enantiomer produced at the step of asymmetric des...
To confirm this issue, kinetic resolution of racemic-6 was performed with catalyst 12b because 12b is almost as effective as 11 in the asymmetric desymmetrization of 5 (Table 1, entry 5 and entry 7). Treatment of rac-6 with 0.7 equiv of isobutyric anhydride in the presence of 5 mol % of 12b gave (1R,2S)-6 in 67% ee at 53% conversion (Scheme 4). This clearly indicates that the (1S,2R)-isomer reacts faster than the (1R,2S)-isomer in the acylation catalyzed by 12b.
![[1860-5397-8-203-i4]](/bjoc/content/inline/1860-5397-8-203-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Acylative kinetic resolution of racemic-6 with catalyst 12b.
Scheme 4: Acylative kinetic resolution of racemic-6 with catalyst 12b.
The results in Table 1 suggest that the amide carbonyl groups at C(2) and C(5) of the pyrrolidine ring of the catalysts 8–14 would play the key role in asymmetric acylation. This is due to the fact that the amide carbonyl group is the common structural subunit among these chiral PPY catalysts. We chose catalyst 11, which possesses the simplest n-hexyl amide side chain, and examined the effect of the secondary amide linkage by comparing the performance of the asymmetric desymmetrization with that shown by the analogous catalysts possessing the corresponding tertiary amide- or ester linkage, 15 or 16, respectively (Table 4, entries 1–3). Much diminished enantioselectivity (13% ee) was observed in the asymmetric desymmetrization of 5 with isobutyric anhydride in the presence of catalyst 15 with the tertiary amide linkage (Table 4, entry 2). Similarly, catalyst 16 with the ester linkage was found to be far less effective (13% ee) than 11 (Table 4, entry 3) in the asymmetric acylation. These results indicate that the secondary amide linkage in 11 is essential for the high efficiency of the asymmetric acylation. The superior property of 11 compared to 16 as an asymmetric acylation catalyst could be ascribed to the stronger Lewis basicity of the amide carbonyl group than that of the ester carbonyl group (donor number of amides > donor number of esters). However, the reasons for the poorer efficiency of catalyst 15 with a tertiary amide linkage compared with catalyst 11 with a secondary amide linkage are unclear (see also Figure 3). We then examined the effects of the C2-symmetric structure of catalysts 11 and 12a by comparing the corresponding mono-functionalized chiral PPY catalysts 17 and 18 [10], respectively (Table 4, entry 4 and entry 5). Catalyst 17 was found to be slightly less effective than 11 in the asymmetric desymmetrization of 5 to give the monoacylate in 76% ee (Table 4, entry 4). Catalyst 18 gave monoacylate 6 in diminished ee (41% ee) in the acylative desymmetrization of 5 (Table 4, entry 5 vs entry 6). The corresponding (1R,2S)-6 was obtained in each case. These results imply that a C2-symmetric structure in catalysts is responsible for the higher efficiency in the asymmetric acylation.
Table 4: Effects of side chain linkage and C2-symmetric structure of catalysts on the asymmetric desymmetrization of 5.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-203-i9.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalystb | 6:7:Recovery of 5c (%) | ee of 6 (%)d,e |
|---|---|---|---|
| 1 | 11 | 75:23:3 | 87 |
| 2 | 15 | 79:15:6 | 13 |
| 3 | 16 | 65:24:11 | 13 |
| 4 | 17 | 70:26:4 | 76 |
| 5 | 18 | 61:34:5 | 41 |
| 6 | 12a | 70:25:5 | 74 |
aReactions were run at a substrate concentration of 0.2 M.
bStructures of catalysts
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-203-i10.svg?max-width=637&scale=1.18182) .
.
cYields determined by 1H NMR with dibenzyl ether as an internal standard. dDetermined by GC analysis with a chiral stationary phase, beta-DEX 225. e(1R,2S)-Isomer was obtained in each case.
Asymmetric desymmetrization of meso-1,3-cyclohexanediol
We have reported that catalyst 3 promoted the acylative asymmetric desymmetrization of meso-1,3-cyclohexanediol (19) to give 20 in 52% ee and 48% yield [11]. Here, C2-symmetric chiral PPY catalysts were examined for this asymmetric transformation (Table 5). Treatment of 19 with isobutyric anhydride in the presence of catalyst 4 at 0 °C gave monoacylate 20 in 48% ee and 66% yield (Table 5, entry 1). The corresponding reaction at −40 °C did not improve the enantioselectivity (Table 5, entry 2). The attempted asymmetric desymmetrization of 19 at 20 °C promoted by catalysts 11, 12a, 12b, and 14 resulted in the formation of the monoacylate in 19–31% ee and 48–69% yield (Table 5, entries 3, 5–7). Lowering the temperature of acylation of 19 in the presence of 11 did not improve the enantioselectivity (Table 5, entry 3 vs entry 4). The lack of temperature effects may indicate that the hydrogen-bonding interaction between the catalyst and the substrate may not significantly be involved in the process of enantioselective acylation of 19 in the presence of catalysts 4 and 11.
Table 5: The asymmetric desymmetrization of meso-1,3-cyclohexanediol (19) with C2-symmetric chiral PPY catalysts.a
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-203-i11.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Temp. (°C) | Time (h) | 20:21:Recovery of 19 (%)b | ee of 20 (%)c,d |
|---|---|---|---|---|---|
| 1 | 4 | 0 | 12 | 66:15:17 | 48 |
| 2 | 4 | –40 | 48 | 26: 2:72 | 50 |
| 3 | 11 | 20 | 4 | 69:24:7 | 31 |
| 4 | 11 | –40 | 48 | 44:10:44 | 25 |
| 5 | 12a | 20 | 4 | 48:43:9 | 27 |
| 6 | 12b | 20 | 4 | 49:30:21 | 20 |
| 7 | 14 | 20 | 4 | 62:33:6 | 19 |
aReactions were run at a substrate concentration of 0.2 M. bYields determined by 1H NMR with dibenzyl ether as an internal standard. cDetermined by GC analysis with a chiral stationary phase, beta-DEX 225. dThe absolute configuration of 20 was not determined.
Asymmetric desymmetrization of meso-2,3-butanediol and meso-hydrobenzoin
The asymmetric desymmetrization of meso-2,3-butanediol (22a) and meso-hydrobenzoin (22b) were examined (Table 6). Treatment of 22a with isobutyric anhydride in the presence of 5 mol % of catalyst 8a at 20 °C for 4 h gave monoacylate 23a in 53% ee and 78% yield (Table 6, entry 1). Catalysts 10, 11, 12b, and 14 were also examined for asymmetric desymmetrization of 22a (Table 6, entries 2–8). These catalysts are almost equally effective in the asymmetric desymmetrization of 22a at 20 °C to give monoacylate 23a in 57–66% ee and 72–78% yield (Table 6, entries 2, 3, 5, and 6). As observed in the asymmetric desymmetrization of 5, the acylation of 22a at a lower temperature gave better selectivity. The catalytic enantioselective acylation of 22a with isobutyric anhydride in the presence of 11 or 14 at −60 to −65 °C gave monoacylate 23a in 87% ee (72% yield) or 92% ee (61% yield), respectively (Table 6, entries 4 and 8) [20] . The higher enantioselectivity was found to be associated with the higher mono/diacylation ratio in the asymmetric acylation of 22a promoted by 14 (Table 6, entries 6–8). (Notice: Enantioenriched 23a gradually undergoes partial racemization when it is kept as a CHCl3 solution probably via intramolecular acyl migration: e.g., from 88% ee to 71% ee after 168 h.) The asymmetric desymmetrization of meso-hydrobenzoin (22b) was examined. Treatment of 22b with isobutyric anhydride in the presence of catalysts 8a, 8b, and 12b at 20 °C gave 23b in 19–40% ee in 54–64% yield (Table 6, entries 9–11). Significant amounts of the diacylate were also formed (22–25% yield) together with the recovery of the unreacted material (11–21%). In these transformations, the low enantioselectivity is associated with the low mono/diacylation ratio, which was also observed in the asymmetric desymmetrization of meso-1,3-cyclohexanediol (19, Table 5). The asymmetric desymmetrization of meso-1,2-cyclopentanediol (22c) was also examined using catalysts 4 and 25 [18], the corresponding octyl ester analogues of 8a and 12a, respectively (Table 6, entries 12–14). The acylation of 22c with isobutyric anhydride in the presence of 4 in chloroform at −20 °C gave monoacylate 23c as a racemate in 85% yield (Table 6, entry 12). Similarly, racemic 23c was obtained by the reaction of 22c with isobutyric anhydride in the presence of 25, either in chloroform or in toluene (Table 6, entries 13 and 14).
Table 6: The asymmetric desymmetrization of meso-2,3-butanediol (22a), meso-hydrobenzoin (22b) and meso-1,2-cyclopentanediol (22c).a
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-203-i12.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Substrate | Catalystb | Temp. (°C) | Time (h) | 23:24:Recovery of 22 (%)c | ee of 23 (%) |
|---|---|---|---|---|---|---|
| 1 | 22a | 8a | 20 | 4 | 78:11:11 | 53d,e |
| 2 | 22a | 10 | 20 | 4 | 77:16:7 | 62d,e |
| 3 | 22a | 11 | 20 | 4 | 78:13:9 | 66d,e |
| 4f | 22a | 11 | −60 | 24 | 72:7:21 | 87d,e |
| 5 | 22a | 12b | 20 | 4 | 72:6:22 | 61d,e |
| 6 | 22a | 14 | 20 | 24 | 73:18:9 | 57d,e |
| 7 | 22a | 14 | −40 | 24 | 82:4:14 | 85d,e |
| 8f | 22a | 14 | −65 | 24 | 61:<1:39 | 92d,e |
| 9 | 22b | 8a | 20 | 4 | 64:25:11 | 40g,h |
| 10 | 22b | 8b | 20 | 4 | 63:22:15 | 23g,h |
| 11 | 22b | 12b | 20 | 4 | 54:25:21 | 19g,h |
| 12 | 22c | 4 | −20 | 6 | 85:12:3 | ~0i |
| 13 | 22c | 25 | −20 | 4 | 73:27:0 | ~0i |
| 14j | 22c | 25 | −20 | 4 | 67:33:0 | ~0i |
aReactions were run at a substrate concentration of 0.2 M.
bStructure of catalyst 25:
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-203-i13.svg?max-width=637&scale=1.18182)
cYields determined by 1H NMR with dibenzyl ether as an internal standard. dEe was determined by GC analysis with a chiral stationary phase, beta-DEX 225. e(2R,3S)-Isomer was obtained. fData quoted from reference [20]. gEe was determined by HPLC analysis with a chiral stationary phase, Chiralcel OJ (iPrOH:hexane = 5:95, flow 0.5 mL min−1, tR = 35, 51 min). hThe absolute configuration was not determined. iEe of the corresponding benzoate, which was determined by HPLC analysis with a chiral stationary phase, Chiralcel AS (iPrOH/hexane 1:99, flow 0.2 mL min−1, tR = 43, 49 min). jRun in toluene.
Mechanistic implication
Several characteristic phenomena were observed in the asymmetric desymmetrization of meso-diols promoted by C2-symmetric chiral PPY catalysts. (1) Substrate specificity: Meso-1,2-cyclohexanediol (5) and meso-2,3-butanediol (22a) (matched substrates) gave high enantioselectivity, while meso-1,3-cyclohexanediol (19), meso-hydrobenzoin (22b), and meso-1,2-cyclopentanediol (22c) (mismatched substrates) gave poor enantioselectivity in the asymmetric desymmetrization. (2) A higher enantioselectivity was observed in the reactions of matched substrate 5 in the solvents of the lower polarity (Table 2, entries 1–5). (3) A higher enantioselectivity and a higher mono/diacylation ratio were observed in the acylation of the matched substrates at the lower temperatures (Table 2, entries 6–9; Table 6, entries 3, 4, 6–8). These phenomena suggest that the enantioselective acylation of the matched substrates proceeds in an accelerative manner via hydrogen-bonding interaction between the catalyst and the substrate. A possible model for the transition state assembly for the enantioselective acylation of meso-1,2-cyclohexanediol (5) catalyzed by 11 is shown in Figure 2. A chiral acylpyridinium ion generated from 11 and isobutyric anhydride is expected to be the reactive intermediate which is responsible for the asymmetric acylation. The most stable conformer A of the acylpyridinium ion was generated by a molecular modeling search (AMBER* force field with the GB/SA solvation model for chloroform using MacroModel V 9.0 (50,000 steps MCMM)) and shown in Figure 2a and Figure 2b. Since the amide carbonyl groups at C(2) and C(5) seem to play the key role in the asymmetric desymmetrization of 5 to give (1R,2S)-6 (Table 1 and Table 4), we assume that the amide carbonyl group would serve as a hydrogen-bond acceptor and the non-reacting OH of 5 as a hydrogen-bond donor. A possible approach of substrate 5 to A is shown in Figure 2c and Figure 2d. In the case where a hydrogen bond between the amide carbonyl group and an axial-OH at the (S)-chiral center of 5 is formed, an equatorial-OH at the (R)-chiral center locates in the close proximity to the reactive carbonyl group without any unfavorable steric interaction, resulting in the selective acylation of the hydroxy group at the (R)-chiral center to give (1R,2S)-6. On the other hand, there may be other possible modes of the approach of 5 to A. They involve hydrogen-bonding interaction between the amide carbonyl group of A and (1) an equatorial-OH at the (S)-chiral center of 5, (2) an axial-OH at the (R)-chiral center of 5, or (3) an equatorial-OH at the (R)-chiral center of 5. The first one would give (1R,2S)-6, while the latter two would give (1S,2R)-6. In these cases, however, unfavorable steric interaction is expected based on our molecular modeling study. It is also anticipated that an axial-OH may be the better hydrogen-bond donor than an equatorial-OH, according to the reported higher acidity of the axial hydroxy groups of cyclohexane derivatives [39]. An alternative model for the transition-state assembly is shown in Figure 3 where the amide NH group of A serves as a hydrogen-bond donor and the non-reacting OH of substrate 5 as a hydrogen-bond acceptor. This model could explain the difference between effective catalyst 11 and ineffective catalysts 15 and 16 (Table 4, entry 1 vs entries 2 and 3). However, the calculated distance between the amide NH group and the reactive amide carbonyl group of A seems too long (8.10 Å) for the accommodation of the 1,2-diol substructure (calculated distance between two oxygen atoms of the hydroxy groups: 2.66 Å). It is also difficult to find the reasons for the preferable acylation of the hydroxy group at the (R)-chiral center of 5 from this model. We prefer the model shown in Figure 2, however, the model in Figure 3 cannot be eliminated.
![[1860-5397-8-203-2]](/bjoc/content/figures/1860-5397-8-203-2.svg?scale=1.8&max-width=1024&background=FFFFFF)
Figure 2: A hypothetical model for the transition-state assembly of the asymmetric acylation of 5 promoted by catalyst 11. Front view a) and side view b) of the calculated structure of acylpyridinium ion A. Front view c) and side view d) of the possible modes for the substrate approach to A.
Figure 2: A hypothetical model for the transition-state assembly of the asymmetric acylation of 5 promoted by...
![[1860-5397-8-203-3]](/bjoc/content/figures/1860-5397-8-203-3.svg?scale=1.7&max-width=1024&background=FFFFFF)
Figure 3: An alternative model for the transition state assembly of the asymmetric acylation of 5 promoted by catalyst 11. a) A possible mode of the substrate approach to acylpyridinium ion A, and b) the calculated structure of A.
Figure 3: An alternative model for the transition state assembly of the asymmetric acylation of 5 promoted by...
Conclusion
We have developed an organocatalytic method for the acylative asymmetric desymmetrization of meso-diols. Highly enantioselective desymmetrization of meso-1,2-cyclohexanediol and meso-2,3-butanediol (matched substrates) was achieved while low to moderate enantioselectivity was observed in the asymmetric desymmetrization of meso-1,3-cyclohexanediol, meso-hydrobenzoin, and meso-1,2-cyclopentanediol (mismatched substrates). Organocatalytic enantioselective acylation of the matched substrates was proposed to proceed via hydrogen-bonding interaction between the catalyst and the substrate.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds, copies of 1H NMR and 13C NMR. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Vedejs, E.; Daugulis, O.; Diver, S. T. J. Org. Chem. 1996, 61, 430–432. doi:10.1021/jo951661v
Return to citation in text: [1] [2] -
Wurz, R. P. Chem. Rev. 2007, 107, 5570–5595. doi:10.1021/cr068370e
Return to citation in text: [1] -
Müller, C. E.; Schreiner, P. R. Angew. Chem., Int. Ed. 2011, 50, 6012–6042. doi:10.1002/anie.201006128
Return to citation in text: [1] -
Höfle, G.; Steglich, W. Synthesis 1972, 619–621. doi:10.1055/s-1972-21955
Return to citation in text: [1] -
Vedejs, E.; Chen, X. J. Am. Chem. Soc. 1996, 118, 1809–1810. doi:10.1021/ja953631f
Return to citation in text: [1] -
Spivey, A. C.; Maddaford, A.; Fekner, T.; Redgrave, A. J.; Frampton, C. S. J. Chem. Soc., Perkin Trans. 1 2000, 3460–3468. doi:10.1039/b004704j
Return to citation in text: [1] -
Naraku, G.; Shimomoto, N.; Nanamoto, T.; Inanaga, J. Enantiomer 2000, 5, 135–136.
Return to citation in text: [1] -
Kawabata, T.; Nagato, M.; Takasu, K.; Fuji, K. J. Am. Chem. Soc. 1997, 119, 3169–3170. doi:10.1021/ja963275g
Return to citation in text: [1] [2] [3] -
Kawabata, T.; Yamamoto, K.; Momose, Y.; Yoshida, H.; Nagaoka, Y.; Fuji, K. Chem. Commun. 2001, 2700–2701. doi:10.1039/b108753c
Return to citation in text: [1] [2] -
Kawabata, T.; Stragies, R.; Fukaya, T.; Fuji, K. Chirality 2003, 15, 71–76. doi:10.1002/chir.10166
Return to citation in text: [1] [2] [3] -
Kawabata, T.; Stragies, R.; Fukaya, T.; Nagaoka, Y.; Schedel, H.; Fuji, K. Tetrahedron Lett. 2003, 44, 1545–1548. doi:10.1016/S0040-4039(03)00021-2
Return to citation in text: [1] [2] [3] [4] -
Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Stragies, R. Synthesis 2008, 747–753. doi:10.1055/s-2008-1032176
Return to citation in text: [1] [2] -
Ueda, Y.; Muramatsu, W.; Mishiro, K.; Furuta, T.; Kawabata, T. J. Org. Chem. 2009, 74, 8802–8805. doi:10.1021/jo901569v
Return to citation in text: [1] [2] -
Yoshida, K.; Furuta, T.; Kawabata, T. Tetrahedron Lett. 2010, 51, 4830–4832. doi:10.1016/j.tetlet.2010.07.036
Return to citation in text: [1] -
Muramatsu, W.; Mishiro, K.; Ueda, Y.; Furuta, T.; Kawabata, T. Eur. J. Org. Chem. 2010, 827–831. doi:10.1002/ejoc.200901393
Return to citation in text: [1] [2] -
Yoshida, K.; Furuta, T.; Kawabata, T. Angew. Chem., Int. Ed. 2011, 50, 4888–4892. doi:10.1002/anie.201100700
Return to citation in text: [1] [2] -
Yoshida, K.; Shigeta, T.; Furuta, T.; Kawabata, T. Chem. Commun. 2012, 48, 6981–6983. doi:10.1039/c2cc32525j
Return to citation in text: [1] [2] -
Sammakia, T.; Hurley, T. B. J. Org. Chem. 1999, 64, 4652–4664. doi:10.1021/jo982281n
Return to citation in text: [1] -
Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Duhamel, L.; Herman, T. Tetrahedron Lett. 1985, 26, 3099–3102. doi:10.1016/S0040-4039(00)98628-3
Return to citation in text: [1] -
Ruble, J. C.; Tweddell, J.; Fu, G. C. J. Org. Chem. 1998, 63, 2794–2795. doi:10.1021/jo980183w
Return to citation in text: [1] -
Oriyama, T.; Imai, K.; Sano, T.; Hosoya, T. Tetrahedron Lett. 1998, 39, 3529–3532. doi:10.1016/S0040-4039(98)00523-1
Return to citation in text: [1] -
Oriyama, T.; Hosoya, T.; Sano, T. Heterocycles 2000, 52, 1065–1069. doi:10.3987/COM-99-S133
Return to citation in text: [1] -
Spivey, A. C.; Zhu, F.; Mitchell, M. B.; Davey, S. G.; Jarvest, L. R. J. Org. Chem. 2003, 68, 7379–7385. doi:10.1021/jo034603f
Return to citation in text: [1] -
Mizuta, S.; Sadamori, M.; Fujimoto, T.; Yamamoto, I. Angew. Chem., Int. Ed. 2003, 42, 3383–3385. doi:10.1002/anie.200250719
Return to citation in text: [1] -
Vedejs, E.; Daugulis, O.; Tuttle, N. J. Org. Chem. 2004, 69, 1389–1392. doi:10.1021/jo030279c
Return to citation in text: [1] -
Kündig, E. P.; Lomberget, T.; Bragg, R.; Poulard, C.; Bernardinelli, G. Chem. Commun. 2004, 1548–1549. doi:10.1039/B404006F
Return to citation in text: [1] -
Yamada, S.; Misono, T.; Iwai, Y. Tetrahedron Lett. 2005, 46, 2239–2242. doi:10.1016/j.tetlet.2005.02.019
Return to citation in text: [1] -
Mizuta, S.; Tsuzuki, T.; Fujimoto, T.; Yamamoto, I. Org. Lett. 2005, 7, 3633–3635. doi:10.1021/ol051129m
Return to citation in text: [1] -
Yamada, S.; Misono, T.; Iwai, Y.; Masumizu, A.; Akiyama, Y. J. Org. Chem. 2006, 71, 6872–6880. doi:10.1021/jo060989t
Return to citation in text: [1] -
Kündig, E. P.; Garcia, A. E.; Lomberget, T.; Bernardinelli, G. Angew. Chem., Int. Ed. 2006, 45, 98–101. doi:10.1002/anie.200502588
Return to citation in text: [1] -
Maki, B. E.; Chan, A.; Phillips, E. M.; Scheidt, K. A. Org. Lett. 2007, 9, 371–374. doi:10.1021/ol062940f
Return to citation in text: [1] -
Birman, V. B.; Jiang, H.; Li, X. Org. Lett. 2007, 9, 3237–3240. doi:10.1021/ol071064i
Return to citation in text: [1] -
Kündig, E. P.; Garcia, A. E.; Lomberget, T.; Garcia, P. P.; Romanens, P. Chem. Commun. 2008, 3519–3521. doi:10.1039/b808268e
Return to citation in text: [1] -
Müller, C. E.; Zell, D.; Shreiner, P. R. Chem.–Eur. J. 2009, 15, 9647–9650. doi:10.1002/chem.200901711
Return to citation in text: [1] -
Maki, B. E.; Chan, A.; Phillips, E. M.; Scheidt, K. A. Tetrahedron 2009, 65, 3102–3109. doi:10.1016/j.tet.2008.10.033
Return to citation in text: [1] -
Cao, J.-L.; Qu, J. J. Org. Chem. 2010, 75, 3663–3670. doi:10.1021/jo100435f
Return to citation in text: [1] -
Majumdar, T. K.; Clairet, F.; Tabet, J.-C.; Cooks, R. G. J. Am. Chem. Soc. 1992, 114, 2897–2903. doi:10.1021/ja00034a021.
Return to citation in text: [1]
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 39. | Majumdar, T. K.; Clairet, F.; Tabet, J.-C.; Cooks, R. G. J. Am. Chem. Soc. 1992, 114, 2897–2903. doi:10.1021/ja00034a021. |
| 1. | Vedejs, E.; Daugulis, O.; Diver, S. T. J. Org. Chem. 1996, 61, 430–432. doi:10.1021/jo951661v |
| 19. | Sammakia, T.; Hurley, T. B. J. Org. Chem. 1999, 64, 4652–4664. doi:10.1021/jo982281n |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 13. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Stragies, R. Synthesis 2008, 747–753. doi:10.1055/s-2008-1032176 |
| 8. | Kawabata, T.; Nagato, M.; Takasu, K.; Fuji, K. J. Am. Chem. Soc. 1997, 119, 3169–3170. doi:10.1021/ja963275g |
| 9. | Kawabata, T.; Yamamoto, K.; Momose, Y.; Yoshida, H.; Nagaoka, Y.; Fuji, K. Chem. Commun. 2001, 2700–2701. doi:10.1039/b108753c |
| 10. | Kawabata, T.; Stragies, R.; Fukaya, T.; Fuji, K. Chirality 2003, 15, 71–76. doi:10.1002/chir.10166 |
| 11. | Kawabata, T.; Stragies, R.; Fukaya, T.; Nagaoka, Y.; Schedel, H.; Fuji, K. Tetrahedron Lett. 2003, 44, 1545–1548. doi:10.1016/S0040-4039(03)00021-2 |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 13. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Stragies, R. Synthesis 2008, 747–753. doi:10.1055/s-2008-1032176 |
| 14. | Ueda, Y.; Muramatsu, W.; Mishiro, K.; Furuta, T.; Kawabata, T. J. Org. Chem. 2009, 74, 8802–8805. doi:10.1021/jo901569v |
| 15. | Yoshida, K.; Furuta, T.; Kawabata, T. Tetrahedron Lett. 2010, 51, 4830–4832. doi:10.1016/j.tetlet.2010.07.036 |
| 16. | Muramatsu, W.; Mishiro, K.; Ueda, Y.; Furuta, T.; Kawabata, T. Eur. J. Org. Chem. 2010, 827–831. doi:10.1002/ejoc.200901393 |
| 17. | Yoshida, K.; Furuta, T.; Kawabata, T. Angew. Chem., Int. Ed. 2011, 50, 4888–4892. doi:10.1002/anie.201100700 |
| 18. | Yoshida, K.; Shigeta, T.; Furuta, T.; Kawabata, T. Chem. Commun. 2012, 48, 6981–6983. doi:10.1039/c2cc32525j |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 4. | Höfle, G.; Steglich, W. Synthesis 1972, 619–621. doi:10.1055/s-1972-21955 |
| 5. | Vedejs, E.; Chen, X. J. Am. Chem. Soc. 1996, 118, 1809–1810. doi:10.1021/ja953631f |
| 6. | Spivey, A. C.; Maddaford, A.; Fekner, T.; Redgrave, A. J.; Frampton, C. S. J. Chem. Soc., Perkin Trans. 1 2000, 3460–3468. doi:10.1039/b004704j |
| 7. | Naraku, G.; Shimomoto, N.; Nanamoto, T.; Inanaga, J. Enantiomer 2000, 5, 135–136. |
| 1. | Vedejs, E.; Daugulis, O.; Diver, S. T. J. Org. Chem. 1996, 61, 430–432. doi:10.1021/jo951661v |
| 21. | Duhamel, L.; Herman, T. Tetrahedron Lett. 1985, 26, 3099–3102. doi:10.1016/S0040-4039(00)98628-3 |
| 22. | Ruble, J. C.; Tweddell, J.; Fu, G. C. J. Org. Chem. 1998, 63, 2794–2795. doi:10.1021/jo980183w |
| 23. | Oriyama, T.; Imai, K.; Sano, T.; Hosoya, T. Tetrahedron Lett. 1998, 39, 3529–3532. doi:10.1016/S0040-4039(98)00523-1 |
| 24. | Oriyama, T.; Hosoya, T.; Sano, T. Heterocycles 2000, 52, 1065–1069. doi:10.3987/COM-99-S133 |
| 25. | Spivey, A. C.; Zhu, F.; Mitchell, M. B.; Davey, S. G.; Jarvest, L. R. J. Org. Chem. 2003, 68, 7379–7385. doi:10.1021/jo034603f |
| 26. | Mizuta, S.; Sadamori, M.; Fujimoto, T.; Yamamoto, I. Angew. Chem., Int. Ed. 2003, 42, 3383–3385. doi:10.1002/anie.200250719 |
| 27. | Vedejs, E.; Daugulis, O.; Tuttle, N. J. Org. Chem. 2004, 69, 1389–1392. doi:10.1021/jo030279c |
| 28. | Kündig, E. P.; Lomberget, T.; Bragg, R.; Poulard, C.; Bernardinelli, G. Chem. Commun. 2004, 1548–1549. doi:10.1039/B404006F |
| 29. | Yamada, S.; Misono, T.; Iwai, Y. Tetrahedron Lett. 2005, 46, 2239–2242. doi:10.1016/j.tetlet.2005.02.019 |
| 30. | Mizuta, S.; Tsuzuki, T.; Fujimoto, T.; Yamamoto, I. Org. Lett. 2005, 7, 3633–3635. doi:10.1021/ol051129m |
| 31. | Yamada, S.; Misono, T.; Iwai, Y.; Masumizu, A.; Akiyama, Y. J. Org. Chem. 2006, 71, 6872–6880. doi:10.1021/jo060989t |
| 32. | Kündig, E. P.; Garcia, A. E.; Lomberget, T.; Bernardinelli, G. Angew. Chem., Int. Ed. 2006, 45, 98–101. doi:10.1002/anie.200502588 |
| 33. | Maki, B. E.; Chan, A.; Phillips, E. M.; Scheidt, K. A. Org. Lett. 2007, 9, 371–374. doi:10.1021/ol062940f |
| 34. | Birman, V. B.; Jiang, H.; Li, X. Org. Lett. 2007, 9, 3237–3240. doi:10.1021/ol071064i |
| 35. | Kündig, E. P.; Garcia, A. E.; Lomberget, T.; Garcia, P. P.; Romanens, P. Chem. Commun. 2008, 3519–3521. doi:10.1039/b808268e |
| 36. | Müller, C. E.; Zell, D.; Shreiner, P. R. Chem.–Eur. J. 2009, 15, 9647–9650. doi:10.1002/chem.200901711 |
| 37. | Maki, B. E.; Chan, A.; Phillips, E. M.; Scheidt, K. A. Tetrahedron 2009, 65, 3102–3109. doi:10.1016/j.tet.2008.10.033 |
| 38. | Cao, J.-L.; Qu, J. J. Org. Chem. 2010, 75, 3663–3670. doi:10.1021/jo100435f |
| 2. | Wurz, R. P. Chem. Rev. 2007, 107, 5570–5595. doi:10.1021/cr068370e |
| 3. | Müller, C. E.; Schreiner, P. R. Angew. Chem., Int. Ed. 2011, 50, 6012–6042. doi:10.1002/anie.201006128 |
| 11. | Kawabata, T.; Stragies, R.; Fukaya, T.; Nagaoka, Y.; Schedel, H.; Fuji, K. Tetrahedron Lett. 2003, 44, 1545–1548. doi:10.1016/S0040-4039(03)00021-2 |
| 11. | Kawabata, T.; Stragies, R.; Fukaya, T.; Nagaoka, Y.; Schedel, H.; Fuji, K. Tetrahedron Lett. 2003, 44, 1545–1548. doi:10.1016/S0040-4039(03)00021-2 |
| 17. | Yoshida, K.; Furuta, T.; Kawabata, T. Angew. Chem., Int. Ed. 2011, 50, 4888–4892. doi:10.1002/anie.201100700 |
| 10. | Kawabata, T.; Stragies, R.; Fukaya, T.; Fuji, K. Chirality 2003, 15, 71–76. doi:10.1002/chir.10166 |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 9. | Kawabata, T.; Yamamoto, K.; Momose, Y.; Yoshida, H.; Nagaoka, Y.; Fuji, K. Chem. Commun. 2001, 2700–2701. doi:10.1039/b108753c |
| 8. | Kawabata, T.; Nagato, M.; Takasu, K.; Fuji, K. J. Am. Chem. Soc. 1997, 119, 3169–3170. doi:10.1021/ja963275g |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 14. | Ueda, Y.; Muramatsu, W.; Mishiro, K.; Furuta, T.; Kawabata, T. J. Org. Chem. 2009, 74, 8802–8805. doi:10.1021/jo901569v |
| 16. | Muramatsu, W.; Mishiro, K.; Ueda, Y.; Furuta, T.; Kawabata, T. Eur. J. Org. Chem. 2010, 827–831. doi:10.1002/ejoc.200901393 |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 12. | Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Uruno, Y.; Schedel, H. J. Am. Chem. Soc. 2007, 129, 12890–12895. doi:10.1021/ja074882e |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 18. | Yoshida, K.; Shigeta, T.; Furuta, T.; Kawabata, T. Chem. Commun. 2012, 48, 6981–6983. doi:10.1039/c2cc32525j |
| 10. | Kawabata, T.; Stragies, R.; Fukaya, T.; Fuji, K. Chirality 2003, 15, 71–76. doi:10.1002/chir.10166 |
| 11. | Kawabata, T.; Stragies, R.; Fukaya, T.; Nagaoka, Y.; Schedel, H.; Fuji, K. Tetrahedron Lett. 2003, 44, 1545–1548. doi:10.1016/S0040-4039(03)00021-2 |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
| 8. | Kawabata, T.; Nagato, M.; Takasu, K.; Fuji, K. J. Am. Chem. Soc. 1997, 119, 3169–3170. doi:10.1021/ja963275g |
| 20. | Furuta, T.; Kawabata, T. Chiral DMAP-Type Catalysts for Acyl-Transfer Reactions. In Asymmetric Organocatalysis 1: Lewis Base and Lewis Acid Catalysts; List, B., Ed.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, New York, 2012; p 518. |
© 2012 Schedel et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)