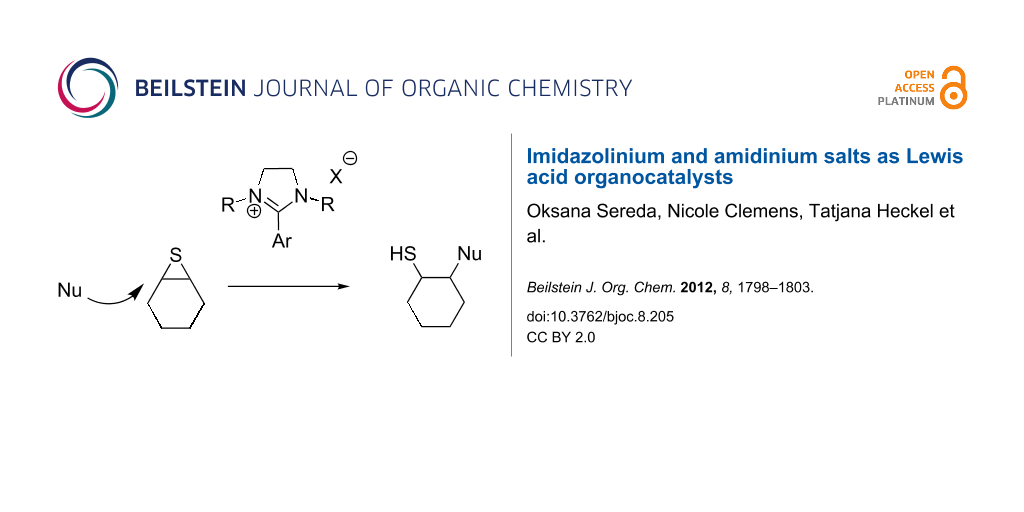
Abstract
The application of imidazolinium and amidinium salts as soft Lewis acid organocatalysts is described. These salts were suitable catalysts for the activation of unsaturated thioesters in a Diels–Alder reaction and in the ring opening of thiiranes and epoxides. The products were isolated in good yields. The mild catalysts did not cause desulfurization of the products containing a thiol or thiocarbonyl group.

Graphical Abstract
Introduction
Salts with melting points below 100 °C are known as ionic liquids and are often used as novel solvents for reactions and electrochemical processes [1]. Several of these solvents can contribute to the research field of “green chemistry” [2]. The most common used ionic liquids are based on imidazolium cations. Next to their application in catalytic reactions [3-6], they are also capable of catalyzing reactions themselves, either in substoichiometric amounts or as reaction medium due to hydrogen-bond activation of the protons of the imidazolium cation [7-10] and other variables, such as π-orbital and charge–charge interactions [8-10].
Recently, we applied saturated imidazolium salts with an aryl substituent at the C(2) position as catalysts for the aza-Diels–Alder reaction [11,12]. These catalysts do not activate the substrate through hydrogen bonding, but instead, like other carbon-cation based catalysts, through their positive center [8-10,13-15], and belong also to the field of organocatalysis [10,16,17].
Taking the soft Lewis acidic character of imidazolinium salts into consideration, we were interested to apply these salts in a Diels–Alder reaction with ethyl crotonthioate as dienophile. Due to the low electronegativity of sulfur it would be difficult to catalyze this reaction with hydrogen-bond activation catalysts [18]. The utilization of the soft Lewis basic sulfur groups in this reaction is rare. Often mercury Lewis acids are applied [19-22] to activate the sulfur group and there is also the risk of desulfurization [23].
The only known asymmetric Diels–Alder reaction with ethyl crotonthioate (1) and cyclopentadiene (2) in the presence of optically pure 2,2'-dimercurio-1,1'-binaphthyl compound 3 gave the product 4 in 44% yield with 58% ee (Scheme 1) [20].
![[1860-5397-8-205-i1]](/bjoc/content/inline/1860-5397-8-205-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diels–Alder reaction with ethyl crotonthioate (1) and cyclopentadiene (2).
Scheme 1: Diels–Alder reaction with ethyl crotonthioate (1) and cyclopentadiene (2).
Results and Discussion
Since product 4 has a low boiling point and proved difficult to purify on a small scale, the model reaction shown in Scheme 2 was evaluated. The α,β-unsaturated thioester 5 was prepared from ethyl cinnamate and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide (Lawesson’s reagent) [22]. The cycloaddition of ethyl thionocinnamate (5) with 1.5 equiv of cyclopentadiene (2) was performed in different solvents, at varied temperatures, by using a broad range of catalysts, which are summarized in Table 1.
![[1860-5397-8-205-i2]](/bjoc/content/inline/1860-5397-8-205-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Diels–Alder reaction catalysed with imidazolinium salts.
Scheme 2: Diels–Alder reaction catalysed with imidazolinium salts.
Table 1: Diels–Alder reaction with 10 mol % catalyst according to Scheme 2. Reactions with CH2Cl2 were carried out in a pressure vessel.
| entry | solvent | catalyst | temp [°C] | time [d] | yield %a (endo:exo) |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | Hg(OAc)2 | −10–45 | 4 | 0 |
| 2 | CH2Cl2 | BF3.Et2O | −10–45 | 4 | 90 (99:1) |
| 3 | CH2Cl2 | TMSOTf | −10–45 | 4 | 55 (99:1) |
| 4 | CH2Cl2 | 7 | 25–45 | 4 | 8 (13:1) |
| 5 | toluene | 7 | 25–110 | 4 | 0 |
| 6 | dioxane | 7 | 25–110 | 4 | 6 (3:1) |
| 7 | CH2Cl2 | 8 | 25–45 | 4 | 5 (9:1) |
| 8 | CH2Cl2 | 9 | 0–45 | 4 | 12 (14:1) |
| 9 | CH2Cl2 | 10 | −10–45 | 4 | 69 (99:1) |
| 10 | CH2Cl2 | 11 | 0–45 | 3 | 66 (99:1) |
aIsolated yields.
First a few metal-based Lewis acids were applied in the reaction. BF3•Et2O is known to activate also sulfur carbonyl groups [24] and gave the product in 90% yield, while TMSOTf gave the product in 55% yield. In both cases the endo product was the major product. The reaction was followed by TLC and the temperature was gradually increased from –10 to 45 °C over 4 days in ca. 10 °C steps.
Next, imidazolinium salt 7 [25] was used in the reaction. The formation of the product was observed at 45 °C; however, the product was isolated only in a low yield of 8%. In toluene no reaction was observed even under reflux. In dioxane under reflux a low yield of 6% was obtained with a low endo/exo ratio of (3:1). Salt 8 [25] and 9 [25] gave similar poor results as salt 7 [25]. Finally, the bis-imidazolinium salt 10 [12] gave the product in 69% yield with an endo/exo ratio comparable to BF3•Et2O. The reaction started slowly at 10 °C. In addition, the similar salt 11 [12] gave the product in a comparable yield of 66%. Although in all cases the product was racemic, the results show that bis-imidazolinium salts can be applied as Lewis acid organocatalysts to activate a thiocarbonyl group.
Next, the behavior of these salts in the ring opening of a thiirane was explored. No activity was observed with salt 9 in the ring opening with aniline. Therefore, the more active bis-imidazolinium salt 10 was applied with thiirane 12 and aniline in CH2Cl2. A yield of 18% was obtained. However, in the absence of a solvent the yield increased to 98% after 16 h (Scheme 3). In the absence of a catalyst a yield of 12% was obtained under neat conditions. Product 13 was obtained in all reactions as racemate in the error range of the HPLC measurements.
![[1860-5397-8-205-i3]](/bjoc/content/inline/1860-5397-8-205-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, several salts were explored in the ring opening of cyclohexene epoxide with aniline (Scheme 4, Table 2). A reaction in CH2Cl2 in the absence of the catalyst gave a yield of 10% after 24 h (Table 2, entry 1).
![[1860-5397-8-205-i4]](/bjoc/content/inline/1860-5397-8-205-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The camphor based salt 16 gave a yield of 24%, while bis-imidazolinium salt 17 resulted in a yield of 60% (Table 2, entries 2 and 3). The bis-imidazolinium salts 10 and 11 gave lower yields compared to 17. However, when the reaction was carried out in the absence of a solvent, the yield increased with salts 10 and 11 to 98 and 96%, respectively (Table 2, entries 6 and 7).
Table 2: Ring opening of epoxide 15 with aniline and 10 mol % salt.
| entry | solvent | catalyst | time | yield %a |
|---|---|---|---|---|
| 1 | CH2Cl2 | – | 24 h | 10 |
| 2 | CH2Cl2 | 16 | 24 h | 24 |
| 3 | CH2Cl2 | 17 | 48 h | 60 |
| 4 | CH2Cl2 | 10 | 48 h | 34 |
| 5 | CH2Cl2 | 11 | 48 h | 48 |
| 6 | neat | 10 | 24 h | 98 |
| 7 | neat | 11 | 24 h | 96 |
| 8 | neat | – | 24 h | 0 |
| 9 | CH2Cl2 | 18 | 48 h | 12 |
| 10 | CH2Cl2 | 19 | 24 h | 78 |
| 11 | CH2Cl2 | 20 | 24 h | 38 |
| 12 | CH2Cl2 | 21 | 6 h | 99 |
| 13 | toluene | – | 2 h | 0 |
| 14 | toluene | 21 | 2 h | 89 |
aIsolated yields.
Compared to sulfur, oxygen is a good hydrogen-bond acceptor. Hence, also imidazolinium salts with a C(2)H unit were applied in the reaction. Here, an activation of the epoxide can be also possible through hydrogen bonding next to the direct interaction with the positively charged NCN center. While salt 18 displayed very low catalytic activity, salt 19 gave the product in 78% yield (Table 2, entry 10). This phenomenon may be explained by the better delocalization of the positive charge in the planar sp2-centered imidazoline scaffold in salt 18. Meanwhile the special geometry of 7-membered 1,3-diazepinium cations such as 19 does not allow the planar conformation to be kept, thus the positive charge is less delocalized over the NCN atoms (Scheme 4) [26-29]. Salt 16, incorporating a six-membered ring as the smallest ring, displayed a catalytic activity between salt 18 and 19. Salt 20, with a larger steric environment around the amidinium unit next to the nitrogen atoms, gave a yield of only 38% (Table 2, entry 11). On the other hand salt 21 gave the product in 99% yield after 6 h (Table 2, entry 12). By changing the solvent from dichloromethane to toluene the reaction time was even further reduced with salt 21, and the product was isolated in 89% yield (Table 2, entry 14). In all cases product 15 was racemic in the error range of the HPLC measurements.
Salt 17 was prepared according to Scheme 5. The synthetic route involved a tetraamine formation via amidation under neat conditions and reduction with LAH, furnishing the product in 98% yield over two steps. Next, the aminal formation under neat conditions was performed. The aminal was further oxidized to a bromide salt by N-bromoacetamide in dimethoxyethane. The bromide counter anion was further efficiently replaced by a tetrakis(3,5-bis(trifluoromethyl)phenyl)borate anion providing the salt 17 by anion metathesis.
![[1860-5397-8-205-i5]](/bjoc/content/inline/1860-5397-8-205-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of bis-imidazolium salt 17.
Scheme 5: Synthesis of bis-imidazolium salt 17.
The 7-membered 1,3-diazepinium salts were prepared according to Scheme 6. Salts 19 and 20 were prepared in a standard way by treatment with triethyl orthoformate in the presence of ammonium tetrafluoroborate followed by anion metathesis in 33 and 29% yield, respectively.
![[1860-5397-8-205-i6]](/bjoc/content/inline/1860-5397-8-205-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of amidinium salt 21.
Scheme 6: Synthesis of amidinium salt 21.
Due to the low yield, salt 21 was prepared through a different route. First amine 27 [30] was transformed with formaldehyde to the aminal 28. The latter could be oxidized to the corresponding bromide salt, which was transformed directly by anion metathesis to the salt 21.
Conclusion
It was possible to prepare new metal-free Lewis acids containing bis-imidazolinium cations and investigate the salts as organocatalysts. Although with enantiopure chiral salts no enantiomeric excess was observed, it was shown for the first time that these salts can interact with thiocarbonyl groups and thiiranes in order to activate these substrates. In addition, it was found that 7-membered 1,3-diazepinium cations are good catalysts for the ring opening of epoxides and that these salts are more reactive than imidazolinium salts.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 205.7 KB | Download |
References
-
Wasserscheid, P.; Welton, T., Eds. Ionic Liquids in Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008; Vol. 1–2.
Return to citation in text: [1] -
Rogers, R. D.; Seddon, K. R., Eds. Ionic Liquids as Green Solvents: Progress and Prospects; American Chemical Society: Washington, DC, 2003.
Return to citation in text: [1] -
Zhao, D.; Wu, M.; Kou, Y.; Min, E. Catal. Today 2002, 74, 157. doi:10.1016/S0920-5861(01)00541-7
Return to citation in text: [1] -
Baudequin, C.; Baudoux, J.; Levillain, J.; Cahard, D.; Gaumont, A.-C.; Plaquevent, J.-C. Tetrahedron: Asymmetry 2003, 14, 3081. doi:10.1016/S0957-4166(03)00596-2
Return to citation in text: [1] -
Jain, N.; Kumar, A.; Chauhan, S.; Chauhan, S. M. S. Tetrahedron 2005, 61, 1015. doi:10.1016/j.tet.2004.10.070
Return to citation in text: [1] -
Pârvulescu, V. I.; Hardacre, C. Chem. Rev. 2007, 107, 2615. doi:10.1021/cr050948h
Return to citation in text: [1] -
Aggarwal, A.; Lancaster, N. L.; Sethi, A. R.; Welton, T. Green Chem. 2002, 4, 517. doi:10.1039/b206472c
Return to citation in text: [1] -
Vidiš, A.; Ohlin, C. A.; Laurenczy, G.; Küsters, E.; Sedelmeier, G.; Dyson, P. J. Adv. Synth. Catal. 2005, 347, 266. doi:10.1002/adsc.200404301
Return to citation in text: [1] [2] [3] -
Tsuzuki, S.; Tokuda, H.; Mikami, M. Phys. Chem. Chem. Phys. 2007, 9, 4780. doi:10.1039/b707419k
Return to citation in text: [1] [2] [3] -
Sereda, O.; Tabasum, S.; Wilhelm, R. Top. Curr. Chem. 2010, 291, 349. doi:10.1007/128_2008_17
See for a review.
Return to citation in text: [1] [2] [3] [4] -
Jurčík, V.; Wilhelm, R. Org. Biomol. Chem. 2005, 3, 239. doi:10.1039/b415023f
Return to citation in text: [1] -
Jurčík, V.; Wilhelm, R. Tetrahedron: Asymmetry 2006, 17, 801. doi:10.1016/j.tetasy.2006.02.021
Return to citation in text: [1] [2] [3] -
Mukaiyama, T.; Yanagisawa, M.; Iida, D.; Hachiya, I. Chem. Lett. 2000, 606. doi:10.1246/cl.2000.606
Return to citation in text: [1] -
Chen, C.-T.; Chao, S.-D.; Yen, K.-C.; Chen, C. H.; Chou, I.-C.; Hon, S.-W. J. Am. Chem. Soc. 1997, 119, 11341. doi:10.1021/ja970900o
Return to citation in text: [1] -
Chen, C.-T.; Chao, S.-D.; Yen, K.-C. Synlett 1998, 924. doi:10.1055/s-1998-1815
Return to citation in text: [1] -
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2001, 40, 3726. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D
See for a review.
Return to citation in text: [1] -
Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley-VCH: Weinheim, Germany, 2005.
See for a review.
Return to citation in text: [1] -
Wittkopp, A.; Schreiner, P. Chem.–Eur. J. 2003, 9, 407. doi:10.1002/chem.200390042
Return to citation in text: [1] -
Lopez, P.; Oh, T. Tetrahedron Lett. 2000, 41, 2313. doi:10.1016/S0040-4039(00)00157-X
Return to citation in text: [1] -
Oh, T.; Lopez, P.; Reilly, M. Eur. J. Org. Chem. 2000, 2901. doi:10.1002/1099-0690(200008)2000:16<2901::AID-EJOC2901>3.0.CO;2-0
Return to citation in text: [1] [2] -
Lee, H.; Diaz, M.; Hawthorne, M. F. Tetrahedron Lett. 1999, 40, 7651. doi:10.1016/S0040-4039(99)01582-8
Return to citation in text: [1] -
Alper, H.; Brandes, D. A. Organometallics 1991, 10, 2457. doi:10.1021/om00053a058
Return to citation in text: [1] [2] -
Wuest, J. D.; Zacharie, B. J. Am. Chem. Soc. 1985, 107, 6121. doi:10.1021/ja00307a058
Return to citation in text: [1] -
Braddock, D. C.; Brown, J. M.; Guiry, P. J. J. Chem. Soc., Chem. Commun. 1993, 1244.
Return to citation in text: [1] -
Clemens, N.; Sereda, O.; Wilhelm, R. Org. Biomol. Chem. 2006, 4, 2285. doi:10.1039/b601104g
Return to citation in text: [1] [2] [3] [4] -
Alder, R. W.; Blake, M. E.; Oliva, J. M. J. Phys. Chem. A 1999, 103, 11200. doi:10.1021/jp9934228
Return to citation in text: [1] -
Magill, A. M.; Cavell, K. J.; Yates, B. F. J. Am. Chem. Soc. 2004, 126, 8717. doi:10.1021/ja038973x
Return to citation in text: [1] -
Scarborough, C. C.; Popp, B. V.; Guzei, I. A.; Stahl, S. S. J. Organomet. Chem. 2005, 690, 6143. doi:10.1016/j.jorganchem.2005.08.022
Return to citation in text: [1] -
Iglesias, M.; Beetstra, D. J.; Knight, J. C.; Ooi, L.-L.; Stasch, A.; Coles, S.; Male, L.; Hursthouse, M. B.; Cavell, K. J.; Dervisi, A.; Fallis, I. A. Organometallics 2008, 27, 3279. doi:10.1021/om800179t
Return to citation in text: [1] -
Angelovski, G.; Keränen, M. D.; Eilbracht, P. Tetrahedron: Asymmetry 2005, 16, 1919. doi:10.1016/j.tetasy.2005.03.035
Return to citation in text: [1]
| 12. | Jurčík, V.; Wilhelm, R. Tetrahedron: Asymmetry 2006, 17, 801. doi:10.1016/j.tetasy.2006.02.021 |
| 25. | Clemens, N.; Sereda, O.; Wilhelm, R. Org. Biomol. Chem. 2006, 4, 2285. doi:10.1039/b601104g |
| 25. | Clemens, N.; Sereda, O.; Wilhelm, R. Org. Biomol. Chem. 2006, 4, 2285. doi:10.1039/b601104g |
| 1. | Wasserscheid, P.; Welton, T., Eds. Ionic Liquids in Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008; Vol. 1–2. |
| 8. | Vidiš, A.; Ohlin, C. A.; Laurenczy, G.; Küsters, E.; Sedelmeier, G.; Dyson, P. J. Adv. Synth. Catal. 2005, 347, 266. doi:10.1002/adsc.200404301 |
| 9. | Tsuzuki, S.; Tokuda, H.; Mikami, M. Phys. Chem. Chem. Phys. 2007, 9, 4780. doi:10.1039/b707419k |
| 10. |
Sereda, O.; Tabasum, S.; Wilhelm, R. Top. Curr. Chem. 2010, 291, 349. doi:10.1007/128_2008_17
See for a review. |
| 25. | Clemens, N.; Sereda, O.; Wilhelm, R. Org. Biomol. Chem. 2006, 4, 2285. doi:10.1039/b601104g |
| 7. | Aggarwal, A.; Lancaster, N. L.; Sethi, A. R.; Welton, T. Green Chem. 2002, 4, 517. doi:10.1039/b206472c |
| 8. | Vidiš, A.; Ohlin, C. A.; Laurenczy, G.; Küsters, E.; Sedelmeier, G.; Dyson, P. J. Adv. Synth. Catal. 2005, 347, 266. doi:10.1002/adsc.200404301 |
| 9. | Tsuzuki, S.; Tokuda, H.; Mikami, M. Phys. Chem. Chem. Phys. 2007, 9, 4780. doi:10.1039/b707419k |
| 10. |
Sereda, O.; Tabasum, S.; Wilhelm, R. Top. Curr. Chem. 2010, 291, 349. doi:10.1007/128_2008_17
See for a review. |
| 25. | Clemens, N.; Sereda, O.; Wilhelm, R. Org. Biomol. Chem. 2006, 4, 2285. doi:10.1039/b601104g |
| 3. | Zhao, D.; Wu, M.; Kou, Y.; Min, E. Catal. Today 2002, 74, 157. doi:10.1016/S0920-5861(01)00541-7 |
| 4. | Baudequin, C.; Baudoux, J.; Levillain, J.; Cahard, D.; Gaumont, A.-C.; Plaquevent, J.-C. Tetrahedron: Asymmetry 2003, 14, 3081. doi:10.1016/S0957-4166(03)00596-2 |
| 5. | Jain, N.; Kumar, A.; Chauhan, S.; Chauhan, S. M. S. Tetrahedron 2005, 61, 1015. doi:10.1016/j.tet.2004.10.070 |
| 6. | Pârvulescu, V. I.; Hardacre, C. Chem. Rev. 2007, 107, 2615. doi:10.1021/cr050948h |
| 22. | Alper, H.; Brandes, D. A. Organometallics 1991, 10, 2457. doi:10.1021/om00053a058 |
| 2. | Rogers, R. D.; Seddon, K. R., Eds. Ionic Liquids as Green Solvents: Progress and Prospects; American Chemical Society: Washington, DC, 2003. |
| 24. | Braddock, D. C.; Brown, J. M.; Guiry, P. J. J. Chem. Soc., Chem. Commun. 1993, 1244. |
| 18. | Wittkopp, A.; Schreiner, P. Chem.–Eur. J. 2003, 9, 407. doi:10.1002/chem.200390042 |
| 23. | Wuest, J. D.; Zacharie, B. J. Am. Chem. Soc. 1985, 107, 6121. doi:10.1021/ja00307a058 |
| 30. | Angelovski, G.; Keränen, M. D.; Eilbracht, P. Tetrahedron: Asymmetry 2005, 16, 1919. doi:10.1016/j.tetasy.2005.03.035 |
| 10. |
Sereda, O.; Tabasum, S.; Wilhelm, R. Top. Curr. Chem. 2010, 291, 349. doi:10.1007/128_2008_17
See for a review. |
| 16. |
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2001, 40, 3726. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D
See for a review. |
| 17. |
Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley-VCH: Weinheim, Germany, 2005.
See for a review. |
| 20. | Oh, T.; Lopez, P.; Reilly, M. Eur. J. Org. Chem. 2000, 2901. doi:10.1002/1099-0690(200008)2000:16<2901::AID-EJOC2901>3.0.CO;2-0 |
| 8. | Vidiš, A.; Ohlin, C. A.; Laurenczy, G.; Küsters, E.; Sedelmeier, G.; Dyson, P. J. Adv. Synth. Catal. 2005, 347, 266. doi:10.1002/adsc.200404301 |
| 9. | Tsuzuki, S.; Tokuda, H.; Mikami, M. Phys. Chem. Chem. Phys. 2007, 9, 4780. doi:10.1039/b707419k |
| 10. |
Sereda, O.; Tabasum, S.; Wilhelm, R. Top. Curr. Chem. 2010, 291, 349. doi:10.1007/128_2008_17
See for a review. |
| 13. | Mukaiyama, T.; Yanagisawa, M.; Iida, D.; Hachiya, I. Chem. Lett. 2000, 606. doi:10.1246/cl.2000.606 |
| 14. | Chen, C.-T.; Chao, S.-D.; Yen, K.-C.; Chen, C. H.; Chou, I.-C.; Hon, S.-W. J. Am. Chem. Soc. 1997, 119, 11341. doi:10.1021/ja970900o |
| 15. | Chen, C.-T.; Chao, S.-D.; Yen, K.-C. Synlett 1998, 924. doi:10.1055/s-1998-1815 |
| 12. | Jurčík, V.; Wilhelm, R. Tetrahedron: Asymmetry 2006, 17, 801. doi:10.1016/j.tetasy.2006.02.021 |
| 11. | Jurčík, V.; Wilhelm, R. Org. Biomol. Chem. 2005, 3, 239. doi:10.1039/b415023f |
| 12. | Jurčík, V.; Wilhelm, R. Tetrahedron: Asymmetry 2006, 17, 801. doi:10.1016/j.tetasy.2006.02.021 |
| 19. | Lopez, P.; Oh, T. Tetrahedron Lett. 2000, 41, 2313. doi:10.1016/S0040-4039(00)00157-X |
| 20. | Oh, T.; Lopez, P.; Reilly, M. Eur. J. Org. Chem. 2000, 2901. doi:10.1002/1099-0690(200008)2000:16<2901::AID-EJOC2901>3.0.CO;2-0 |
| 21. | Lee, H.; Diaz, M.; Hawthorne, M. F. Tetrahedron Lett. 1999, 40, 7651. doi:10.1016/S0040-4039(99)01582-8 |
| 22. | Alper, H.; Brandes, D. A. Organometallics 1991, 10, 2457. doi:10.1021/om00053a058 |
| 26. | Alder, R. W.; Blake, M. E.; Oliva, J. M. J. Phys. Chem. A 1999, 103, 11200. doi:10.1021/jp9934228 |
| 27. | Magill, A. M.; Cavell, K. J.; Yates, B. F. J. Am. Chem. Soc. 2004, 126, 8717. doi:10.1021/ja038973x |
| 28. | Scarborough, C. C.; Popp, B. V.; Guzei, I. A.; Stahl, S. S. J. Organomet. Chem. 2005, 690, 6143. doi:10.1016/j.jorganchem.2005.08.022 |
| 29. | Iglesias, M.; Beetstra, D. J.; Knight, J. C.; Ooi, L.-L.; Stasch, A.; Coles, S.; Male, L.; Hursthouse, M. B.; Cavell, K. J.; Dervisi, A.; Fallis, I. A. Organometallics 2008, 27, 3279. doi:10.1021/om800179t |
© 2012 Sereda et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)