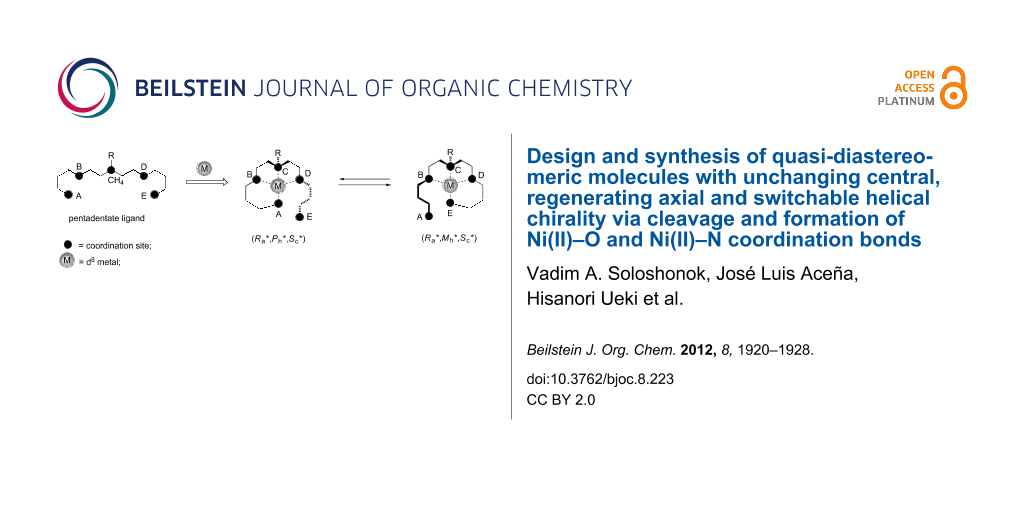
Abstract
We describe herein the design and synthesis of asymmetric, pentadentate ligands, which are able to coordinate to Ni(II) cations leading to quasi-diastereomeric complexes displaying two new elements of chirality: stereogenic axis and helix along with configurational stabilization of the stereogenic center on the nitrogen. Due to the stereocongested structural characteristics of the corresponding Ni(II) complexes, the formation of quasi-diastereomeric products is highly stereoselective providing formation of only two, (Ra*,Mh*,Rc*) and (Ra*,Ph*,Rc*), out of the four possible stereochemical combinations. The reversible quasi-diastereomeric transformation between the products (Ra*,Mh*,Rc*) and (Ra*,Ph*,Rc*) occurs by intramolecular trans-coordination of Ni–NH and Ni–O bonds providing a basis for a chiral switch model.

Graphical Abstract
Introduction
The design and synthesis of organic molecules for which the changing of their three-dimensional structure or function can be predicted, is a fast-growing multidisciplinary field of research [1-13]. In particular, organic compounds that can undergo reversible diastereomeric transformations are considered as the most promising models for the development of molecular switches with nondestructive read out of the optical information [14-18]. Taking into account the issue of fatigue resistance or durability of a potential molecular switch, the newly emerging design strategies make use of the formation and cleavage of metal–ligand coordination bonds as a basis for a chiral conformational switch in the conformationally restricted molecular backbone [19-23]. Here, we would like to describe a new design and synthesis of quasi-diastereomeric molecules with unchanging central, regenerating axial, and switchable helical chirality, through cleavage/formation of Ni(II)–O and Ni(II)–N coordination bonds.
Recently, we introduced a new approach to the design of organic molecules with switchable chirality by simple cleavage and formation of metal–ligand coordination bonds, as a potentially useful and conceptually new model for the development of a new generation of organic chiroptical molecular switches [24,25]. Thus, achiral C2-symmetric pentadentate ligands 1 were able to coordinate with d8 metals [Ni(II) or Pd(II)] to form tetra-coordinated diastereomeric complexes (Ra*,Ph*,Sc*)-2 and (Ra*,Mh*,Rc*)-3, which contain at least three elements of chirality, namely stereogenic center, axis and helix (Scheme 1). The structure of these complexes incorporated two five- and one six-membered ring displaying a square-planar geometry. It is worth mentioning that only two out of the four possible diastereomeric complexes were accessed in a highly stereoselective manner, as a result of the intrinsic steric hindrance associated with the design of achiral ligands 1. In addition, crystallization of the diastereomeric mixtures resulted in the formation of a single diastereomer (Ra*,Ph*,Sc*)-2 or (Ra*,Mh*,Rc*)-3 in the solid state, depending on the nature of the chelating metal (Ni or Pd) employed.
![[1860-5397-8-223-i1]](/bjoc/content/inline/1860-5397-8-223-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previous design of diastereomeric molecules 2 and 3 with switchable chirality starting from achiral, C2-symmetric pentadentate ligands 1.
Scheme 1: Previous design of diastereomeric molecules 2 and 3 with switchable chirality starting from achiral...
In this context, we subsequently envisioned the design and synthesis of structurally related complexes in which the chiral switch will be based on trans-coordination of nonsymmetric ligands (Scheme 2).
![[1860-5397-8-223-i2]](/bjoc/content/inline/1860-5397-8-223-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General design of asymmetric pentadentate ligands 4 and chiroptically switchable quasi-diastereomeric molecules 5 and 6, possessing elements of central, axial and helical chirality.
Scheme 2: General design of asymmetric pentadentate ligands 4 and chiroptically switchable quasi-diastereomer...
The major difference of the general design presented in Scheme 2 with the previously reported models (Scheme 1) is that the starting pentadentate ligand 4 is asymmetric and therefore already possesses the stereogenic center at the coordination site C. It is assumed that the pentadentate ligand 4 upon coordination with d8 metals will give rise to tetra-coordinated square-planar complexes 5 and 6. The presence of coordinated and noncoordinated groups A and E, positioned up or down relative to the metal coordination plane, will create an element of axial chirality, directed through the coordination sites D in complex 5 and B in 6, and the corresponding adjacent methylenes of the chelating five-membered rings. The second, newly created element of chirality may be provided by steric repulsive interactions in the arms BA/DE, which will twist the six-membered chelating ring resulting in a screw sense of helical chirality. The transcoordination motion in these complexes is expected to be controlled in a way that noncoordinated sites, i.e., E in 5 and A in 6, will move up and down, respectively, resulting in the interconversion of complexes 5 and 6. This assumed concurrent and unidirectional motion of arms ED and AB is expected to have the following desired stereochemical consequences: Thus, the axial chirality on the arm CDE in 5 will disappear with simultaneous generation of a new chiral axis on the arm CBA in 6, of the same stereochemical sense. Next, this envisioned mode of transcoordination must proceed also with a loss and regeneration of helical chirality, but, in contrast to the loss/regeneration of axial chirality, with the inversion of the stereochemical sense, providing quasi-diastereomeric relationships between complexes 5 and 6 [26].
As one can expect, these desired structural and stereochemical considerations can be seriously compromised by the presence of central chirality in the starting ligand 4. For instance, application of racemic ligands 4 will give rise to at least four diastereomeric products rendering the designed diastereomeric intertransformation with switchable chirality simply pointless. Furthermore, application of enantiomerically pure ligands 4 can present a problem of mismatched stereochemical preferences between the existing central and newly generated set of axial and helical chirality. On the other hand, if the stereogenic center in ligands 4 is configurationally flexible (can easily undergo (S) to (R) transformation) one may expect the opposite trend, as the combined stereochemical preferences of axial and helical chirality will impose the corresponding matching configuration of the stereogenic center. Thus, the geometric difference between the designed quasi-diastereomers 5 and 6 is that the noncoordinated arm CDE and the substituent R on the stereogenic coordination site C in 5 are on the same side of the metal chelation plane, while in structure of 6 the arm CBA and the substituent R are on the opposite sides. In contrast, the geometric relations between the off-plane six-membered chelate rings, giving rise to helical chirality, and the substituent R are the same in both quasi-diastereomers 5 and 6. Considering these geometric features, one may expect that the actual absolute configuration of the stereogenic center C, determined by the stereochemical priority of the groups A and E, has no importance for the quasi-diastereomeric relationships between 5 and 6. Consequently, there is a good chance that the geometric position of the substituent R, and therefore the relative configuration of the stereogenic center on C, may be efficiently controlled by the matching stereochemical preferences between the axial and helical chirality in the quasi-diastereomers 5 and 6. However, to realize this possibility the stereogenic center C in ligands 4 should be configurationally unstable and easily adaptable to the developing stereochemical environment upon its configurational stabilization in complexes 5 and 6.
Results and Discussion
To build the real molecules to this design, we explored the preparation of imino–carbonyl ligands 13 (Scheme 3) by desymmetrization of achiral carbonyl–carbonyl ligands 12. Compounds 12 were prepared in the framework of our recently described methodology for applying a new generation of nucleophilic glycine equivalents [27-32] to a general asymmetric synthesis of α-amino acids [33-38]. In this manner, modular assembly of achiral C2-symmetric pentadentate ligands 12 was carried out by using three inexpensive and readily available starting structural units. First, the “phenone” modules 7 were reacted [39] with “acid” module 8 to form the corresponding amides 9 in quantitative chemical yield. Application of compounds 9 for bis-alkylation of “amine” module 10 was conducted in two separate steps including isolation and purification of the intermediate products 11. The undesired formation of the corresponding quaternary ammonium salts was not observed [40] and the target ligands 12 were prepared in high overall yield (96%).
![[1860-5397-8-223-i3]](/bjoc/content/inline/1860-5397-8-223-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of imino–carbonyl ligands 13 by desymmetrization of achiral carbonyl–carbonyl ligands 12.
Scheme 3: Preparation of imino–carbonyl ligands 13 by desymmetrization of achiral carbonyl–carbonyl ligands 12...
Surprisingly, we found that the seemingly simple transformation of 12 to 13 can present a significant synthetic problem. Thus, our attempts to prepare the desired mono-imino ligands 13 from compounds 12, using one equivalent of various amines, resulted in the formation of complex mixtures of inseparable products. Possible reasons for this outcome are the presence of two potentially reactive amide groups and the multifunctional nature of compounds 12. In particular, preparation of the most desirable NH-containing (R’ = H) derivatives 13, by reaction of 12 with ammonia or its derivatives, was not possible at all. Modification of the synthetic scheme with installation of the imino functionality on intermediate stages also gave inacceptable results, probably due to hydrolytic liability of the corresponding imino derivatives of, for instance, compounds 9 and 11.
While these unexpected synthetic obstacles can, probably, be overcome with a more extensive search for optimized conditions, we decided to take advantage of our experience in the chemistry of Ni(II)-chelated amino acid Schiff bases [41-48] and to develop an indirect approach for the preparation of asymmetric ligands of type 13. To this goal, we studied the reactions of biscarbonyl compounds 12 with glycine.
As shown in Scheme 4, heating of pentadentate ligands 12 in acetonitrile in the presence of a Ni(II) source [NiCl2 or Ni(NO3)2], glycine and a base, lead to the formation of the quite unusual complexes 14. In a similar manner, by using PdCl2 instead of Ni(II), the Pd-chelated complex 15 was obtained. As it follows from these results, only one carbonyl group in the starting ligands 12 was involved in the formation of the corresponding Schiff base with glycine, similar to the reactivity observed for tridentate ligands with Ni(II) [49-53]. In the 1H and 13C NMR spectra of compounds 14 and 15, resonances of aromatic and aliphatic hydrogens and carbons appear as sharp peaks indicating that the extra bidentate moiety in complexes 14 and 15 acts merely as a substituent and does not compete for coordination with Ni(II). Complexes 14 are red-colored, 15 is yellow, and both are neutral, highly crystalline compounds (mp > 250 °C), good solubility in organic solvents, and can be easily purified by regular column chromatography on SiO2.
![[1860-5397-8-223-i4]](/bjoc/content/inline/1860-5397-8-223-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of complexes 14a,b and 15 by reactions of ligands 12 with glycine.
Scheme 4: Preparation of complexes 14a,b and 15 by reactions of ligands 12 with glycine.
To receive more precise data about the structure of complexes 14 and 15 we performed crystallographic analysis of compound 14a [54] (Figure 1). Quite surprisingly, we found that in the solid state (monoclinic crystal system, space group P21/n) complex 14a has three elements of chirality: stereogenic center [N(2)], axis [N(1)–C(8) bond] and helix (off-plane position of the chelate rings). While these three elements of chirality can give up to four possible diastereomeric combinations, only one diastereomer, as a pair of (Rc,Ra,Ph) and (Sc,Sa,Mh) enantiomers, was found in the crystallographic unit cell. This fact clearly suggested that the observed set of stereochemical preferences is a result of a mutual match between these three elements of chirality giving rise to the most stable diastereomeric stereochemistry.
![[1860-5397-8-223-1]](/bjoc/content/figures/1860-5397-8-223-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystallographic structure of complex 14a.
Figure 1: Crystallographic structure of complex 14a.
The distances of the Ni–O(5) [1.8651(13) Å] and Ni–N(4) [1.8569(16) Å] bonds of the five-membered chelate ring of the glycine Schiff base fragment were relatively similar, while the bonds Ni–N(3) [1.8417(16) Å] and Ni–N(2) [1.9535(16) Å] were noticeably shorter and longer, respectively. The square-planar structure of compound 14a is slightly distorted with angles N(4)–Ni–N(2) of 173.48(7)° and N(3)–Ni–O(5) of 175.15(6)°. This deviation from planarity gives compound 14a a bowl-like shape and renders it inherently chiral. For instance, the torsion angle C(19)–N(3)–C(20)–C(21) of 32.0(3)° sets the element of helical chirality, while the axial chirality is located through the N(1)–C(8) bond with the torsion angle C(9)–N(1)–C(8)–C(7) of −139.5(2)°. The central chirality is located on N(2) as, coordinated to Ni(II), nitrogen is configurationally stable and has four different substituents. As mentioned above, the absorptions of hydrogens and carbons in the NMR spectra, recorded at ambient temperature, are sharp and therefore do not suggest any diastereomeric transformations. Accordingly, one may assume that in CHCl3 solution compounds 14 and 15 also exist in one diastereomeric form possibly stabilized by apical coordination of the amidic carbonyl of the pendent side chain (distance of Ni–O(2) = 2.665 Å).
With complexes 14 and 15 in hand, we next studied the oxidation of 14 as presented in Scheme 5. Formation of the corresponding enolates 16, in the presence of relatively strong bases (KOt-Bu), is an established step in general methods for homologation of the glycine moiety by alkyl halide alkylation [27,50-53], aldol [29,33,36,37] and Michael [28,31,34,35,38,39,43-47] addition reactions. The following oxidation step of the enolates 16 in the presence of atmospheric oxygen, is less studied [48]; however, plausible key transformations can be deduced based on the closely related results on the well-established general oxidation of enolates [55-64] and glycine-derived enolates [65-70], in particular, under similar conditions. Thus, the ionized form of α-hydroxy intermediate 17 can undergo the cleavage of the glycine C–N bond [65,68-70] resulting in the formation of neutral complex 18. Previously, we demonstrated that in situ formed derivatives of type 18, without the pendant side chain, undergo further stabilization through dimerization, giving rise to the corresponding binuclear complexes [48]. In this case, the presence of the bidentate substituent allows for a different, intramolecular stabilization mechanism leading to the formation of target complexes 19/20.
![[1860-5397-8-223-i5]](/bjoc/content/inline/1860-5397-8-223-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Oxidation of enolates 16 and formation of complexes 19 and 20.
Scheme 5: Oxidation of enolates 16 and formation of complexes 19 and 20.
It is interesting to note that the acetophenone module-derived compound 14a gave the expected enolate 16a; however, its further oxidation lead mostly to decomposition, suggesting higher instability of intermediate derivatives of type 17 and 18 [48]. Also, the benzophenone-derived, Pd(II)-containing analogue 15 (Scheme 4) failed to produce the target complexes of type 19/20, pointing to some limitation of this synthetic alternative. Nevertheless, this oxidative pathway allowed for convenient preparation of otherwise inaccessible complexes 19/20 formally derived from asymmetric carbonyl/imine ligands 13 (Scheme 3) and Ni(II). In contrast to typical Ni(II) complexes, such as 14a,b, compounds 19/20 display an unusual dark-brown color. Nevertheless, their high solubility in polar organic solvents allows for their straightforward purification by column chromatography. Initial assignment of the structures of compounds 19/20 by NMR was not possible because of the very fast exchange between Ni–O=C and Ni–NH=C chelation. Thus, due to fast interconversion between compounds 19 and 20, only broad signals were observed in the 1H NMR spectra recorded at ambient temperature. However, variable temperature experiments (−50 °C) produced a notable sharpening in both aromatic and aliphatic protons. Ultimately, elucidation of their structure was carried out by X-ray analysis, which was feasible due to their high crystallinity [54].
We found that in the solid state, compounds 19 and 20 exist as the sole NH–Ni coordinated quasi-diastereomer (Ra*,Mh*,Rc*)-19 (Figure 2), containing a pair of (Ra,Mh,Rc) and (Sa,Ph,Sc) enantiomers in the crystallographic unit cell (triclinic, ![[Graphic 1]](/bjoc/content/inline/1860-5397-8-223-i6.svg?max-width=637&scale=1.18182) space group). Surprisingly, the distance of the Ni–NH bond was found to be the shortest [1.8518(15) Å] in the Ni(II)-coordinated plane, followed by the Ni–N(3) [1.8524(14) Å] of the same five-membered chelate ring. Two other Ni–N(1) [1.8760(14) Å] and Ni(1)–N(2) [1.9076(15) Å] were noticeably longer. The square-planar coordination plane in compound 19 was substantially more distorted, as compared with complex 14a or carbonyl-coordinated analogues 2 and 3 [24,25] (Scheme 1), with the angles N(3)–Ni–N(1) of 171.94(6)° and N(4)–Ni–N(2) of 169.80(6)° rendering it inherently chiral. Consequently, the torsion angle C(24)–N(3)–C(25)–C(26) of 34.4(2)° gives rise to an element of helical chirality in compound 19. The element of axial chirality is generated by the restricted rotation around the N(1)–C(13) bond with the torsion angle C(14)–N(1)–C(13)–C(12) of −119.59(18)°. The third element of chirality, a stereogenic center, is located on N(2).
space group). Surprisingly, the distance of the Ni–NH bond was found to be the shortest [1.8518(15) Å] in the Ni(II)-coordinated plane, followed by the Ni–N(3) [1.8524(14) Å] of the same five-membered chelate ring. Two other Ni–N(1) [1.8760(14) Å] and Ni(1)–N(2) [1.9076(15) Å] were noticeably longer. The square-planar coordination plane in compound 19 was substantially more distorted, as compared with complex 14a or carbonyl-coordinated analogues 2 and 3 [24,25] (Scheme 1), with the angles N(3)–Ni–N(1) of 171.94(6)° and N(4)–Ni–N(2) of 169.80(6)° rendering it inherently chiral. Consequently, the torsion angle C(24)–N(3)–C(25)–C(26) of 34.4(2)° gives rise to an element of helical chirality in compound 19. The element of axial chirality is generated by the restricted rotation around the N(1)–C(13) bond with the torsion angle C(14)–N(1)–C(13)–C(12) of −119.59(18)°. The third element of chirality, a stereogenic center, is located on N(2).
![[1860-5397-8-223-2]](/bjoc/content/figures/1860-5397-8-223-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystallographic structure of complex 19.
Figure 2: Crystallographic structure of complex 19.
Conclusion
In conclusion, we have shown that the newly prepared Ni(II) complexes derived from asymmetric, pentadentate ligands exist as quasi-diastereomers, generating two new elements of chirality (stereogenic axis and helix) to the pre-existing configurationally stable stereogenic nitrogen. The stereoselective formation of only two, (Ra*,Mh*,Rc*) and (Ra*,Ph*,Rc*), out of the four possible stereochemical combinations is most remarkable, arising due to the highly stereocongested structural characteristics of these Ni(II) complexes. The quasi-diastereomeric transformation between products (Ra*,Mh*,Rc*) and (Ra*,Ph*,Rc*) occurs by intramolecular transcoordination of Ni–NH and Ni–O bonds. This transcoordination results in the loss and regeneration of axial and helical chirality with retention and inversion, respectively, of the stereochemical sense. The stereogenic center in this case remains unchanged. As it follows from the crystallographic studies, in the solid state there is an obvious preference for Ni–NH over Ni–O coordination. The flexibility of this modular design, in which the three major “phenone”, “acid” and “amine” modules can be selected by using many structural modifications, provides a very practical tool for the preparation of a wide variety of Ni(II) complexes, which may result in a higher sensitivity to chemical properties such as temperature, solvent or pH of the reaction medium.
Experimental
General
1H and 13C NMR were performed on Varian Unity-300 (299.94 MHz) and Gemini-200 (199.98 MHz) spectrometers with TMS and CDCl3 as internal standards. High-resolution mass spectra (HRMS) were recorded on a JEOL HX110A instrument. Optical rotations were measured on a JASCO P-1010 polarimeter. Melting points (mp) are uncorrected and were obtained in open capillaries. All reagents and solvents, unless otherwise stated, are commercially available and were used as received. Unless otherwise stated, yields refer to isolated yields of products of greater than 95% purity as estimated by 1H and 13C NMR spectrometry. All new compounds were characterized by 1H, 13C NMR and HRMS. For preparation of 12a,b see [25,39].
Synthesis of complexes 14a,b and 15
General procedure. To a suspension of compound 12, glycine (5 equiv), and MX2 (NiCl2, Ni(NO3)2 or PdCl2, 2 equiv) in MeOH, NaOH (7 equiv) was added, and the reaction mixture was stirred at 60–70 °C for 4 h. Then, the reaction mixture was poured over a slurry of ice and 5% AcOH. After complete precipitation, the solid was filtered, washed with water, and dried. The product was purified by silica-gel column chromatography (CHCl3/acetone = 4:1).
Complex 14a (46% yield). Mp 277.2 °C (dec); 1H NMR (300 MHz, CDCl3) δ 2.34 (s, 3H), 2.68 (s, 3H), 2.89 (d, J = 15.4 Hz, 1H), 3.30 (d, J = 16.5 Hz, 1H), 3.83 (d, J = 15.4 Hz, 1H), 3.85 (d, J = 16.2 Hz, 1H), 3.86 (d, J = 18.8 Hz, 1H), 3.94 (d, J = 12.6 Hz, 1H), 4.43 (d, J = 19.0 Hz, 1H), 4.54 (d, J = 12.8 Hz, 1H), 6.92 (ddd, J = 8.30, 7.13, 1.18 Hz, 1H), 7.16–7.24 (m, 2H), 7.30–7.36 (m, 1H), 7.37–7.46 (m, 2H), 7.50 (dd, J = 8.30, 1.46 Hz, 1H), 7.64 (ddd, J = 8.69, 7.13, 1.56 Hz, 1H), 7.93 (dd, J = 8.01, 1.56 Hz, 1H), 8.06 (dd, J = 8.59, 1.17 Hz, 1H), 8.09–8.15 (m, 2H), 8.97 (dd, J = 8.49, 1.17 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 18.9, 28.5, 59.8, 60.0, 60.9, 64.4, 121.2, 121.3, 122.1, 123.3, 124.8, 126.8, 128.7, 128.9, 129.4, 131.4, 131.5, 131.7, 132.1, 135.2, 139.9, 141.4, 166.7, 168.8, 176.1, 176.8, 203.2; HRMS–ESI (m/z): [M + Na]+ calcd for C29H28N4NaNiO5, 593.1311; found, 593.1321.
Complex 14b (83% yield). Mp 259.3 °C (dec); 1H NMR (300 MHz, CDCl3) δ 2.83 (d, J = 16.5 Hz, 1H), 3.37 (d, J = 16.4 Hz, 1H), 3.65 (d, J = 19.8 Hz, 1H), 3.84 (d, J = 12.2 Hz, 1H), 3.88 (d, J = 13.2 Hz, 1H), 3.89 (d, J = 19.8 Hz, 1H), 4.14 (d, J = 12.8 Hz, 1H), 4.70 (d, J = 13.1 Hz, 1H), 6.72 (ddd, J = 8.30, 6.84, 1.27 Hz, 1H), 6.84 (dd, J = 8.20, 1.76 Hz, 1H), 7.00–7.74 (m, 17H), 7.90–8.00 (m, 2H), 8.47 (d, J = 8.79 Hz, 1H), 8.83 (d, J = 7.91 Hz, 1H), 11.1 (bs, 1H); 13C NMR (75 MHz, CDCl3) δ 59.0, 60.5, 61.2, 63.9, 120.7, 122.6, 123.3, 124.2, 125.1, 125.5, 125.9, 126.3, 128.2, 128.9, 129.1, 129.4, 129.4, 129.6, 129.8, 131.1, 132.0, 132.1, 132.5, 133.0, 133.1, 133.9, 134.7, 138.1, 138.7, 142.4, 166.5, 171.0, 176.7, 177.1, 199.2; HRMS–ESI (m/z): [M + H]+ calcd for C39H33N4NiO5, 695.1804; found, 695.1801.
Complex 15 (86% yield). Mp 253.5 °C (dec); 1H NMR (300 MHz, CDCl3): δ 3.77 (d, J = 15.8 Hz, 1H), 4.11 (d, J = 11.6 Hz, 1H), 4.12 (s, 2H), 4.18 (d, J = 11.1 Hz, 1H), 4.30 (d, J = 16.4 Hz, 1H), 4.46 (s, 2H), 6.78 (ddd, J = 8.20, 7.03, 1.17 Hz, 1H), 6.92–7.01 (m, 2H), 7.13–7.22 (m, 2H), 7.25–7.88 (m, 16H), 8.47 (dd, J = 8.79, 1.18 Hz, 1H), 8.58 (dd, J = 8.50, 1.18 Hz, 1H), 11.0 (bs, 1H); 13C NMR (75 MHz, CDCl3) δ 61.1, 63.1, 64.0, 65.3, 121.2, 122.4, 123.4, 123.6, 125.0, 126.0, 126.2, 126.4, 128.3, 128.8, 129.4, 129.5, 129.7, 129.7, 129.9, 131.5, 132.3, 132.7, 133.0, 133.1, 133.8, 134.2, 134.9, 137.9, 138.6, 142.2, 165.0, 169.2, 176.6, 177.8, 198.9; HRMS–ESI (m/z): [M + H]+ calcd for C39H33N4O5Pd, 743.1436; found, 743.1501.
Complexes 19/20. To a solution of 14b (1 equiv) in CH3CN, was added KOt-Bu (4 equiv) at rt and the reaction mixture was stirred at rt under aerobic conditions until the starting compound was consumed completely, as confirmed by TLC. After evaporation of the solvent, water and a calculated amount of 5% AcOH aq. (4 equiv) was added and extracted with CH2Cl2 three times. The combined organic layers were dried over MgSO4, and the product was purified on a short-path flash silica-gel column. Due to the fast exchange between complexes 19/20, NMR spectra were not properly recorded. Mp 264.5 °C (dec); HRMS–ESI (m/z): [M + Na]+ calcd for C37H30N4NaNiO3, 659.1569; found, 659.1547.
Supporting Information
| Supporting Information File 1: NMR spectra of compounds 14b and 19/20. | ||
| Format: PDF | Size: 878.7 KB | Download |
References
-
Feringa, B. L.; Koumura, N.; van Delden, R. A.; ter Wiel, M. K. J. Appl. Phys. A 2002, 75, 301–308. doi:10.1007/s003390201338
Return to citation in text: [1] -
de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515–1566. doi:10.1021/cr960386p
Return to citation in text: [1] -
Rambidi, N. G. Microelectron. Eng. 2003, 69, 485–500. doi:10.1016/S0167-9317(03)00337-X
Return to citation in text: [1] -
Janek, J. Nat. Mater. 2009, 8, 88–89. doi:10.1038/nmat2364
Return to citation in text: [1] -
Fukui, M.; Mori, T.; Inoue, Y.; Rathore, R. Org. Lett. 2007, 9, 3977–3980. doi:10.1021/ol701639u
Return to citation in text: [1] -
Deng, J.; Song, N.; Zhou, Q.; Su, Z. Org. Lett. 2007, 9, 5393–5396. doi:10.1021/ol701822u
Return to citation in text: [1] -
Mori, T.; Inoue, Y. J. Phys. Chem. A 2005, 109, 2728–2740. doi:10.1021/jp044917m
Return to citation in text: [1] -
Shie, T.-L.; Lin, C.-H.; Lin, S.-L.; Yang, D.-Y. Eur. J. Org. Chem. 2007, 4831–4836. doi:10.1002/ejoc.200700426
Return to citation in text: [1] -
Collin, J.-P.; Kern, J.-M.; Raehm, L.; Sauvage, J.-P. Metallo-Rotaxanes and Catenanes as Redox Switches: Towards Molecular Machines and Motors. In Molecular Switches; Feringa, B. L., Ed.; Wiley-VCH: Weinheim, Germany, 2001; pp 249–280.
Return to citation in text: [1] -
Stoddart, J. F.; Colquhoun, H. M. Tetrahedron 2008, 64, 8231–8263. doi:10.1016/j.tet.2008.06.035
Return to citation in text: [1] -
Kay, E. R.; Leigh, D. A. Pure Appl. Chem. 2008, 80, 17–29. doi:10.1351/pac200880010017
Return to citation in text: [1] -
Kim, Y.-H.; Goddard, W. A., III. J. Phys. Chem. C 2007, 111, 4831–4837. doi:10.1021/jp065302n
Return to citation in text: [1] -
Saha, S.; Stoddart, J. F. Chem. Soc. Rev. 2007, 36, 77–92. doi:10.1039/b607187b
Return to citation in text: [1] -
Irie, M., Ed. Photochromism: Memories and Switches. Chem. Rev. 2000, 100, 1683–1890. doi:10.1021/cr980068l
Return to citation in text: [1] -
Pijper, D.; Jongejan, M. G. M.; Meetsma, A.; Feringa, B. L. J. Am. Chem. Soc. 2008, 130, 4541–4552. doi:10.1021/ja711283c
Return to citation in text: [1] -
Geertsema, E. M.; Hoen, R.; Meetsma, A.; Feringa, B. L. Eur. J. Org. Chem. 2006, 3596–3605. doi:10.1002/ejoc.200600280
Return to citation in text: [1] -
Zheng, J.; Qiao, W.; Wan, X.; Gao, J. P.; Wang, Z. Y. Chem. Mater. 2008, 20, 6163–6168. doi:10.1021/cm8014644
Return to citation in text: [1] -
Wang, Z. Y.; Todd, E. K.; Meng, X. S.; Gao, J. P. J. Am. Chem. Soc. 2005, 127, 11552–11553. doi:10.1021/ja0526721
Return to citation in text: [1] -
Shinkai, S.; Ikeda, M.; Sugasaki, A.; Takeuchi, M. Acc. Chem. Res. 2001, 34, 494–503. doi:10.1021/ar000177y
Return to citation in text: [1] -
Jiang, X.; Lim, Y.-K.; Zhang, B. J.; Opsitnick, E. A.; Baik, M.-H.; Lee, D. J. Am. Chem. Soc. 2008, 130, 16812–16822. doi:10.1021/ja806723e
Return to citation in text: [1] -
Li, Y.; Wang, T.; Liu, M. Soft Matter 2007, 3, 1312–1317. doi:10.1039/b710165a
Return to citation in text: [1] -
Qiu, Y.; Chen, P.; Guo, P.; Li, Y.; Liu, M. Adv. Mater. 2008, 20, 2908–2913. doi:10.1002/adma.200801165
Return to citation in text: [1] -
Canary, J. W.; Zahn, S.; Chiu, Y.-H.; Santos, O.; Liu, J.; Zhu, L. Enantiomer 2000, 5, 397–403.
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Moore, J. L.; Ellis, T. K. J. Am. Chem. Soc. 2007, 129, 3512–3513. doi:10.1021/ja067995r
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090
Return to citation in text: [1] [2] [3] -
The term “quasi-diastereomer” is used by analogy with “quasi-enantiomer”, as defined by Eliel and Wilen: Eliel, E. L.; Wilen, S. H. Stereochemistry of Organic Compunds; Wiley-Interscience: New York, 1994.
Return to citation in text: [1] -
Ellis, T. K.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2005, 46, 941–944. doi:10.1016/j.tetlet.2004.12.050
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K.; Yamada, T.; Ohfune, Y. Tetrahedron Lett. 2005, 46, 1107–1110. doi:10.1016/j.tetlet.2004.12.093
Return to citation in text: [1] [2] -
Ellis, T. K.; Soloshonok, V. A. Synlett 2006, 533–538. doi:10.1055/s-2006-926252
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Chim. Oggi 2008, 26, 51–54.
Return to citation in text: [1] -
Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V. A. Tetrahedron: Asymmetry 2008, 19, 2789–2795. doi:10.1016/j.tetasy.2008.11.036
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929
Return to citation in text: [1] -
Soloshonok, V. A.; Belokon, Y. N.; Kuzmina, N. A.; Maleev, V. I.; Svistunova, N. Y.; Solodenko, V. A.; Kukhar, V. P. J. Chem. Soc., Perkin Trans. 1 1992, 1525–1529. doi:10.1039/P19920001525
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron: Asymmetry 1999, 10, 4265–4269. doi:10.1016/S0957-4166(99)00483-8
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Meervelt, L. V.; Mischenko, N. Tetrahedron Lett. 1997, 38, 4903–4904. doi:10.1016/S0040-4039(97)01054-X
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P. Tetrahedron: Asymmetry 1996, 7, 1547–1550. doi:10.1016/0957-4166(96)00177-2
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Tararov, V. I.; Saveleva, T. F.; Churkina, T. D.; Ikonnikov, N. S.; Kochetkov, K. A.; Orlova, S. A.; Pysarevsky, A. P.; Struchkov, Y. T.; Raevsky, N. I.; Belokon, Y. N. Tetrahedron: Asymmetry 1995, 6, 1741–1756. doi:10.1016/0957-4166(95)00220-J
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Org. Lett. 2000, 2, 747–750. doi:10.1021/ol990402f
Return to citation in text: [1] [2] -
Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198
Return to citation in text: [1] [2] [3] -
Moore, J. L.; Taylor, S. M.; Soloshonok, V. A. ARKIVOC 2005, No. vi, 287–292.
Return to citation in text: [1] -
Ueki, H.; Ellis, T. K.; Martin, C. H.; Soloshonok, V. A. Eur. J. Org. Chem. 2003, 1954–1957. doi:10.1002/ejoc.200200688
Return to citation in text: [1] -
Ueki, H.; Ellis, T. K.; Martin, C. H.; Boettiger, T. U.; Bolene, S. B.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 7104–7107. doi:10.1021/jo0301494
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Angew. Chem., Int. Ed. 2000, 39, 2172–2175. doi:10.1002/1521-3773(20000616)39:12<2172::AID-ANIE2172>3.0.CO;2-0
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 9645–9649. doi:10.1016/S0040-4039(00)01737-8
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 135–139. doi:10.1016/S0040-4039(99)02018-3
Return to citation in text: [1] [2] -
Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V. A. Org. Lett. 2006, 8, 5625–5628. doi:10.1021/ol0623668
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215
Return to citation in text: [1] [2] [3] [4] -
Qiu, W.; Soloshonok, V. A.; Cai, C.; Tang, X.; Hruby, V. J. Tetrahedron 2000, 56, 2577–2582. doi:10.1016/S0040-4020(00)00176-9
Return to citation in text: [1] -
Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Tang, X.; Hruby, V. J.; Meervelt, L. V. Org. Lett. 2001, 3, 341–343. doi:10.1021/ol000330o
Return to citation in text: [1] [2] -
Ellis, T. K.; Martin, C. H.; Tsai, G. M.; Ueki, H.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 6208–6214. doi:10.1021/jo030075w
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Tang, X.; Hruby, V. J. Tetrahedron 2001, 57, 6375–6382. doi:10.1016/S0040-4020(01)00504-X
Return to citation in text: [1] [2] -
CCDC 875510 and 875511 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] [2] -
Christoffers, J.; Baro, A.; Werner, T. Adv. Synth. Catal. 2004, 346, 143–151. doi:10.1002/adsc.200303140
See for a review.
Return to citation in text: [1] -
Rahman, M. T.; Nishino, H. Org. Lett. 2003, 5, 2887–2890. doi:10.1021/ol034939b
Return to citation in text: [1] -
Christoffers, J. J. Org. Chem. 1999, 64, 7668–7669. doi:10.1021/jo9909094
Return to citation in text: [1] -
Christoffers, J.; Werner, T.; Unger, S.; Frey, W. Eur. J. Org. Chem. 2003, 425–431. doi:10.1002/ejoc.200390075
Return to citation in text: [1] -
Christoffers, J.; Werner, T. Synlett 2002, 119–121. doi:10.1055/s-2002-19331
Return to citation in text: [1] -
Arai, T.; Akazome, M.; Ogura, K. Chem. Lett. 1999, 28, 107–108. doi:10.1246/cl.1999.107
Return to citation in text: [1] -
Adam, W.; Cueto, O.; Ehrig, V. J. Org. Chem. 1976, 41, 370–371. doi:10.1021/jo00864a040
Return to citation in text: [1] -
Wasserman, H. H.; Lipshutz, B. H. Tetrahedron Lett. 1975, 16, 1731–1734. doi:10.1016/S0040-4039(00)72245-3
Return to citation in text: [1] -
Acocella, M. R.; Mancheño, O. G.; Bella, M.; Jørgensen, K. A. J. Org. Chem. 2004, 69, 8165–8167. doi:10.1021/jo048655w
Return to citation in text: [1] -
Jefford, C. W.; Currie, J.; Richardson, G. D.; Rossier, J. C. Helv. Chim. Acta 1991, 74, 1239–1246. doi:10.1002/hlca.19910740612
Return to citation in text: [1] -
Ooi, T.; Takeuchi, M.; Ohara, D.; Maruoka, K. Synlett 2001, 1185–1187. doi:10.1055/s-2001-15167
Return to citation in text: [1] [2] -
Bull, S. D.; Davies, S. G.; Garner, A. C.; O’Shea, M. D.; Savory, E. D.; Snow, E. J. J. Chem. Soc., Perkin Trans. 1 2002, 2442–2448. doi:10.1039/B207457P
Return to citation in text: [1] -
Williams, R. M.; Armstrong, R. W.; Dung, J. S. J. Am. Chem. Soc. 1984, 106, 5748–5750. doi:10.1021/ja00331a066
Return to citation in text: [1] -
Garcia-Raso, A.; Deyá, P. M.; Saá, J. M. J. Org. Chem. 1986, 51, 4285–4287. doi:10.1021/jo00372a033
Return to citation in text: [1] [2] -
Easton, C. J.; Eichinger, S. K.; Pitt, M. J. J. Chem. Soc., Chem. Commun. 1992, 1295–1296. doi:10.1039/C39920001295
Return to citation in text: [1] [2] -
Easton, C. J.; Eichinger, S. K.; Pitt, M. J. Tetrahedron 1997, 53, 5609–5616. doi:10.1016/S0040-4020(97)00216-0
Return to citation in text: [1] [2]
| 65. | Ooi, T.; Takeuchi, M.; Ohara, D.; Maruoka, K. Synlett 2001, 1185–1187. doi:10.1055/s-2001-15167 |
| 68. | Garcia-Raso, A.; Deyá, P. M.; Saá, J. M. J. Org. Chem. 1986, 51, 4285–4287. doi:10.1021/jo00372a033 |
| 69. | Easton, C. J.; Eichinger, S. K.; Pitt, M. J. J. Chem. Soc., Chem. Commun. 1992, 1295–1296. doi:10.1039/C39920001295 |
| 70. | Easton, C. J.; Eichinger, S. K.; Pitt, M. J. Tetrahedron 1997, 53, 5609–5616. doi:10.1016/S0040-4020(97)00216-0 |
| 55. |
Christoffers, J.; Baro, A.; Werner, T. Adv. Synth. Catal. 2004, 346, 143–151. doi:10.1002/adsc.200303140
See for a review. |
| 56. | Rahman, M. T.; Nishino, H. Org. Lett. 2003, 5, 2887–2890. doi:10.1021/ol034939b |
| 57. | Christoffers, J. J. Org. Chem. 1999, 64, 7668–7669. doi:10.1021/jo9909094 |
| 58. | Christoffers, J.; Werner, T.; Unger, S.; Frey, W. Eur. J. Org. Chem. 2003, 425–431. doi:10.1002/ejoc.200390075 |
| 59. | Christoffers, J.; Werner, T. Synlett 2002, 119–121. doi:10.1055/s-2002-19331 |
| 60. | Arai, T.; Akazome, M.; Ogura, K. Chem. Lett. 1999, 28, 107–108. doi:10.1246/cl.1999.107 |
| 61. | Adam, W.; Cueto, O.; Ehrig, V. J. Org. Chem. 1976, 41, 370–371. doi:10.1021/jo00864a040 |
| 62. | Wasserman, H. H.; Lipshutz, B. H. Tetrahedron Lett. 1975, 16, 1731–1734. doi:10.1016/S0040-4039(00)72245-3 |
| 63. | Acocella, M. R.; Mancheño, O. G.; Bella, M.; Jørgensen, K. A. J. Org. Chem. 2004, 69, 8165–8167. doi:10.1021/jo048655w |
| 64. | Jefford, C. W.; Currie, J.; Richardson, G. D.; Rossier, J. C. Helv. Chim. Acta 1991, 74, 1239–1246. doi:10.1002/hlca.19910740612 |
| 65. | Ooi, T.; Takeuchi, M.; Ohara, D.; Maruoka, K. Synlett 2001, 1185–1187. doi:10.1055/s-2001-15167 |
| 66. | Bull, S. D.; Davies, S. G.; Garner, A. C.; O’Shea, M. D.; Savory, E. D.; Snow, E. J. J. Chem. Soc., Perkin Trans. 1 2002, 2442–2448. doi:10.1039/B207457P |
| 67. | Williams, R. M.; Armstrong, R. W.; Dung, J. S. J. Am. Chem. Soc. 1984, 106, 5748–5750. doi:10.1021/ja00331a066 |
| 68. | Garcia-Raso, A.; Deyá, P. M.; Saá, J. M. J. Org. Chem. 1986, 51, 4285–4287. doi:10.1021/jo00372a033 |
| 69. | Easton, C. J.; Eichinger, S. K.; Pitt, M. J. J. Chem. Soc., Chem. Commun. 1992, 1295–1296. doi:10.1039/C39920001295 |
| 70. | Easton, C. J.; Eichinger, S. K.; Pitt, M. J. Tetrahedron 1997, 53, 5609–5616. doi:10.1016/S0040-4020(97)00216-0 |
| 1. | Feringa, B. L.; Koumura, N.; van Delden, R. A.; ter Wiel, M. K. J. Appl. Phys. A 2002, 75, 301–308. doi:10.1007/s003390201338 |
| 2. | de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515–1566. doi:10.1021/cr960386p |
| 3. | Rambidi, N. G. Microelectron. Eng. 2003, 69, 485–500. doi:10.1016/S0167-9317(03)00337-X |
| 4. | Janek, J. Nat. Mater. 2009, 8, 88–89. doi:10.1038/nmat2364 |
| 5. | Fukui, M.; Mori, T.; Inoue, Y.; Rathore, R. Org. Lett. 2007, 9, 3977–3980. doi:10.1021/ol701639u |
| 6. | Deng, J.; Song, N.; Zhou, Q.; Su, Z. Org. Lett. 2007, 9, 5393–5396. doi:10.1021/ol701822u |
| 7. | Mori, T.; Inoue, Y. J. Phys. Chem. A 2005, 109, 2728–2740. doi:10.1021/jp044917m |
| 8. | Shie, T.-L.; Lin, C.-H.; Lin, S.-L.; Yang, D.-Y. Eur. J. Org. Chem. 2007, 4831–4836. doi:10.1002/ejoc.200700426 |
| 9. | Collin, J.-P.; Kern, J.-M.; Raehm, L.; Sauvage, J.-P. Metallo-Rotaxanes and Catenanes as Redox Switches: Towards Molecular Machines and Motors. In Molecular Switches; Feringa, B. L., Ed.; Wiley-VCH: Weinheim, Germany, 2001; pp 249–280. |
| 10. | Stoddart, J. F.; Colquhoun, H. M. Tetrahedron 2008, 64, 8231–8263. doi:10.1016/j.tet.2008.06.035 |
| 11. | Kay, E. R.; Leigh, D. A. Pure Appl. Chem. 2008, 80, 17–29. doi:10.1351/pac200880010017 |
| 12. | Kim, Y.-H.; Goddard, W. A., III. J. Phys. Chem. C 2007, 111, 4831–4837. doi:10.1021/jp065302n |
| 13. | Saha, S.; Stoddart, J. F. Chem. Soc. Rev. 2007, 36, 77–92. doi:10.1039/b607187b |
| 26. | The term “quasi-diastereomer” is used by analogy with “quasi-enantiomer”, as defined by Eliel and Wilen: Eliel, E. L.; Wilen, S. H. Stereochemistry of Organic Compunds; Wiley-Interscience: New York, 1994. |
| 28. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K.; Yamada, T.; Ohfune, Y. Tetrahedron Lett. 2005, 46, 1107–1110. doi:10.1016/j.tetlet.2004.12.093 |
| 31. | Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V. A. Tetrahedron: Asymmetry 2008, 19, 2789–2795. doi:10.1016/j.tetasy.2008.11.036 |
| 34. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron: Asymmetry 1999, 10, 4265–4269. doi:10.1016/S0957-4166(99)00483-8 |
| 35. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Meervelt, L. V.; Mischenko, N. Tetrahedron Lett. 1997, 38, 4903–4904. doi:10.1016/S0040-4039(97)01054-X |
| 38. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Org. Lett. 2000, 2, 747–750. doi:10.1021/ol990402f |
| 39. | Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198 |
| 43. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Angew. Chem., Int. Ed. 2000, 39, 2172–2175. doi:10.1002/1521-3773(20000616)39:12<2172::AID-ANIE2172>3.0.CO;2-0 |
| 44. | Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561 |
| 45. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 9645–9649. doi:10.1016/S0040-4039(00)01737-8 |
| 46. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 135–139. doi:10.1016/S0040-4039(99)02018-3 |
| 47. | Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V. A. Org. Lett. 2006, 8, 5625–5628. doi:10.1021/ol0623668 |
| 24. | Soloshonok, V. A.; Ueki, H.; Moore, J. L.; Ellis, T. K. J. Am. Chem. Soc. 2007, 129, 3512–3513. doi:10.1021/ja067995r |
| 25. | Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090 |
| 48. | Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215 |
| 19. | Shinkai, S.; Ikeda, M.; Sugasaki, A.; Takeuchi, M. Acc. Chem. Res. 2001, 34, 494–503. doi:10.1021/ar000177y |
| 20. | Jiang, X.; Lim, Y.-K.; Zhang, B. J.; Opsitnick, E. A.; Baik, M.-H.; Lee, D. J. Am. Chem. Soc. 2008, 130, 16812–16822. doi:10.1021/ja806723e |
| 21. | Li, Y.; Wang, T.; Liu, M. Soft Matter 2007, 3, 1312–1317. doi:10.1039/b710165a |
| 22. | Qiu, Y.; Chen, P.; Guo, P.; Li, Y.; Liu, M. Adv. Mater. 2008, 20, 2908–2913. doi:10.1002/adma.200801165 |
| 23. | Canary, J. W.; Zahn, S.; Chiu, Y.-H.; Santos, O.; Liu, J.; Zhu, L. Enantiomer 2000, 5, 397–403. |
| 27. | Ellis, T. K.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2005, 46, 941–944. doi:10.1016/j.tetlet.2004.12.050 |
| 50. | Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014 |
| 51. | Soloshonok, V. A.; Tang, X.; Hruby, V. J.; Meervelt, L. V. Org. Lett. 2001, 3, 341–343. doi:10.1021/ol000330o |
| 52. | Ellis, T. K.; Martin, C. H.; Tsai, G. M.; Ueki, H.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 6208–6214. doi:10.1021/jo030075w |
| 53. | Soloshonok, V. A.; Tang, X.; Hruby, V. J. Tetrahedron 2001, 57, 6375–6382. doi:10.1016/S0040-4020(01)00504-X |
| 25. | Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090 |
| 39. | Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198 |
| 14. | Irie, M., Ed. Photochromism: Memories and Switches. Chem. Rev. 2000, 100, 1683–1890. doi:10.1021/cr980068l |
| 15. | Pijper, D.; Jongejan, M. G. M.; Meetsma, A.; Feringa, B. L. J. Am. Chem. Soc. 2008, 130, 4541–4552. doi:10.1021/ja711283c |
| 16. | Geertsema, E. M.; Hoen, R.; Meetsma, A.; Feringa, B. L. Eur. J. Org. Chem. 2006, 3596–3605. doi:10.1002/ejoc.200600280 |
| 17. | Zheng, J.; Qiao, W.; Wan, X.; Gao, J. P.; Wang, Z. Y. Chem. Mater. 2008, 20, 6163–6168. doi:10.1021/cm8014644 |
| 18. | Wang, Z. Y.; Todd, E. K.; Meng, X. S.; Gao, J. P. J. Am. Chem. Soc. 2005, 127, 11552–11553. doi:10.1021/ja0526721 |
| 29. | Ellis, T. K.; Soloshonok, V. A. Synlett 2006, 533–538. doi:10.1055/s-2006-926252 |
| 33. | Soloshonok, V. A.; Belokon, Y. N.; Kuzmina, N. A.; Maleev, V. I.; Svistunova, N. Y.; Solodenko, V. A.; Kukhar, V. P. J. Chem. Soc., Perkin Trans. 1 1992, 1525–1529. doi:10.1039/P19920001525 |
| 36. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P. Tetrahedron: Asymmetry 1996, 7, 1547–1550. doi:10.1016/0957-4166(96)00177-2 |
| 37. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Tararov, V. I.; Saveleva, T. F.; Churkina, T. D.; Ikonnikov, N. S.; Kochetkov, K. A.; Orlova, S. A.; Pysarevsky, A. P.; Struchkov, Y. T.; Raevsky, N. I.; Belokon, Y. N. Tetrahedron: Asymmetry 1995, 6, 1741–1756. doi:10.1016/0957-4166(95)00220-J |
| 40. | Moore, J. L.; Taylor, S. M.; Soloshonok, V. A. ARKIVOC 2005, No. vi, 287–292. |
| 49. | Qiu, W.; Soloshonok, V. A.; Cai, C.; Tang, X.; Hruby, V. J. Tetrahedron 2000, 56, 2577–2582. doi:10.1016/S0040-4020(00)00176-9 |
| 50. | Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014 |
| 51. | Soloshonok, V. A.; Tang, X.; Hruby, V. J.; Meervelt, L. V. Org. Lett. 2001, 3, 341–343. doi:10.1021/ol000330o |
| 52. | Ellis, T. K.; Martin, C. H.; Tsai, G. M.; Ueki, H.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 6208–6214. doi:10.1021/jo030075w |
| 53. | Soloshonok, V. A.; Tang, X.; Hruby, V. J. Tetrahedron 2001, 57, 6375–6382. doi:10.1016/S0040-4020(01)00504-X |
| 54. | CCDC 875510 and 875511 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 39. | Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198 |
| 54. | CCDC 875510 and 875511 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 24. | Soloshonok, V. A.; Ueki, H.; Moore, J. L.; Ellis, T. K. J. Am. Chem. Soc. 2007, 129, 3512–3513. doi:10.1021/ja067995r |
| 25. | Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090 |
| 33. | Soloshonok, V. A.; Belokon, Y. N.; Kuzmina, N. A.; Maleev, V. I.; Svistunova, N. Y.; Solodenko, V. A.; Kukhar, V. P. J. Chem. Soc., Perkin Trans. 1 1992, 1525–1529. doi:10.1039/P19920001525 |
| 34. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron: Asymmetry 1999, 10, 4265–4269. doi:10.1016/S0957-4166(99)00483-8 |
| 35. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Meervelt, L. V.; Mischenko, N. Tetrahedron Lett. 1997, 38, 4903–4904. doi:10.1016/S0040-4039(97)01054-X |
| 36. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P. Tetrahedron: Asymmetry 1996, 7, 1547–1550. doi:10.1016/0957-4166(96)00177-2 |
| 37. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Tararov, V. I.; Saveleva, T. F.; Churkina, T. D.; Ikonnikov, N. S.; Kochetkov, K. A.; Orlova, S. A.; Pysarevsky, A. P.; Struchkov, Y. T.; Raevsky, N. I.; Belokon, Y. N. Tetrahedron: Asymmetry 1995, 6, 1741–1756. doi:10.1016/0957-4166(95)00220-J |
| 38. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Org. Lett. 2000, 2, 747–750. doi:10.1021/ol990402f |
| 48. | Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215 |
| 27. | Ellis, T. K.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2005, 46, 941–944. doi:10.1016/j.tetlet.2004.12.050 |
| 28. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K.; Yamada, T.; Ohfune, Y. Tetrahedron Lett. 2005, 46, 1107–1110. doi:10.1016/j.tetlet.2004.12.093 |
| 29. | Ellis, T. K.; Soloshonok, V. A. Synlett 2006, 533–538. doi:10.1055/s-2006-926252 |
| 30. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Chim. Oggi 2008, 26, 51–54. |
| 31. | Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V. A. Tetrahedron: Asymmetry 2008, 19, 2789–2795. doi:10.1016/j.tetasy.2008.11.036 |
| 32. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929 |
| 41. | Ueki, H.; Ellis, T. K.; Martin, C. H.; Soloshonok, V. A. Eur. J. Org. Chem. 2003, 1954–1957. doi:10.1002/ejoc.200200688 |
| 42. | Ueki, H.; Ellis, T. K.; Martin, C. H.; Boettiger, T. U.; Bolene, S. B.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 7104–7107. doi:10.1021/jo0301494 |
| 43. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Angew. Chem., Int. Ed. 2000, 39, 2172–2175. doi:10.1002/1521-3773(20000616)39:12<2172::AID-ANIE2172>3.0.CO;2-0 |
| 44. | Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561 |
| 45. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 9645–9649. doi:10.1016/S0040-4039(00)01737-8 |
| 46. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 135–139. doi:10.1016/S0040-4039(99)02018-3 |
| 47. | Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V. A. Org. Lett. 2006, 8, 5625–5628. doi:10.1021/ol0623668 |
| 48. | Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215 |
| 48. | Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215 |
© 2012 Soloshonok et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)